CNRS UMR 8199, European Genomic Institute for Diabetes (EGID), Institut Pasteur de Lille, University of Lille, Lille, France; Department of Pediatrics, Saint Antoine Pediatric Hospital, Saint Vincent de Paul Hospital, Groupement des Hôpitaux de l'Institut Catholique de Lille (GHICL), Catholic University of Lille, Lille, France.
CNRS UMR 8199, European Genomic Institute for Diabetes (EGID), Institut Pasteur de Lille, University of Lille, Lille, France.
Mol Metab. 2018 Jul;13:1-9. doi: 10.1016/j.molmet.2018.05.005. Epub 2018 May 16.
The molecular diagnosis of extreme forms of obesity, in which accurate detection of both copy number variations (CNVs) and point mutations, is crucial for an optimal care of the patients and genetic counseling for their families. Whole-exome sequencing (WES) has benefited considerably this molecular diagnosis, but its poor ability to detect CNVs remains a major limitation. We aimed to develop a method (CoDE-seq) enabling the accurate detection of both CNVs and point mutations in one step.
CoDE-seq is based on an augmented WES method, using probes distributed uniformly throughout the genome. CoDE-seq was validated in 40 patients for whom chromosomal DNA microarray was available. CNVs and mutations were assessed in 82 children/young adults with suspected Mendelian obesity and/or intellectual disability and in their parents when available (n = 145).
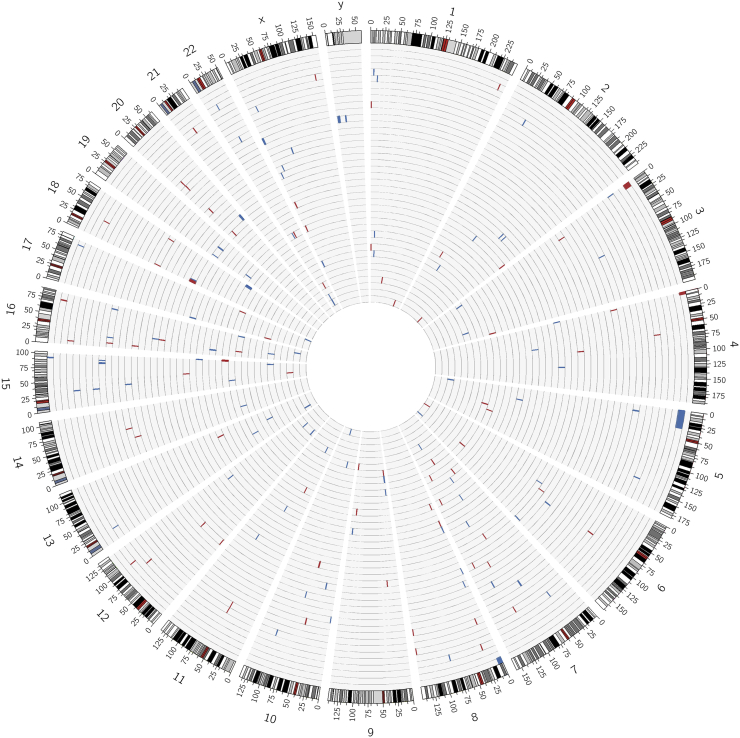
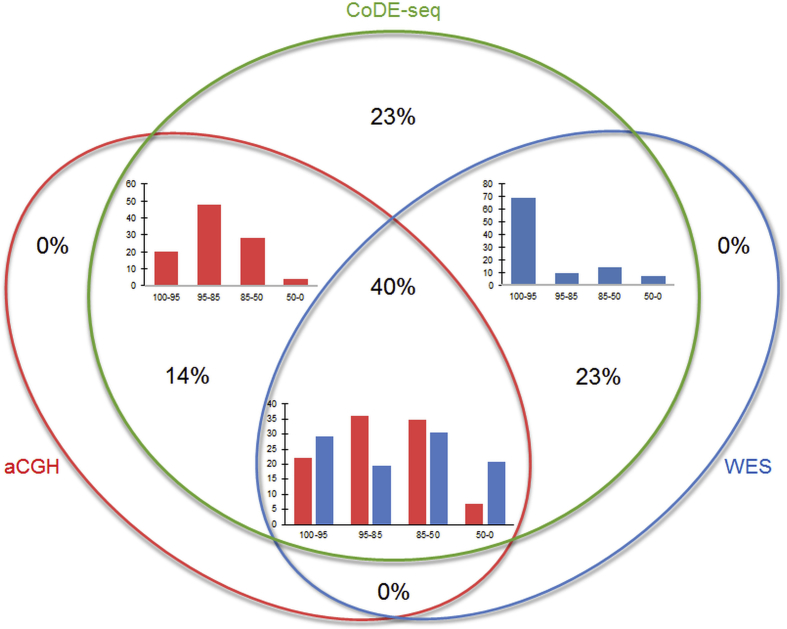
CoDE-seq not only detected all of the 97 CNVs identified by chromosomal DNA microarrays but also found 84 additional CNVs, due to a better resolution. When compared to CoDE-seq and chromosomal DNA microarrays, WES failed to detect 37% and 14% of CNVs, respectively. In the 82 patients, a likely molecular diagnosis was achieved in >30% of the patients. Half of the genetic diagnoses were explained by CNVs while the other half by mutations.
CoDE-seq has proven cost-efficient and highly effective as it avoids the sequential genetic screening approaches currently used in clinical practice for the accurate detection of CNVs and point mutations.
对于肥胖的极端形式,准确检测拷贝数变异(CNVs)和点突变对于患者的最佳治疗和遗传咨询至关重要。全外显子组测序(WES)极大地促进了这种分子诊断,但检测 CNVs 的能力仍然是一个主要限制。我们旨在开发一种方法(CoDE-seq),能够在一步中准确检测 CNVs 和点突变。
CoDE-seq 基于一种扩展的 WES 方法,使用分布在整个基因组中的探针。在 40 名患者中验证了 CoDE-seq,他们的染色体 DNA 微阵列可用。在 82 名疑似孟德尔肥胖症和/或智力障碍的儿童/年轻成人及其父母(n=145)中评估了 CNVs 和突变。
CoDE-seq 不仅检测到了染色体 DNA 微阵列确定的 97 个 CNVs,而且由于分辨率更高,还发现了 84 个额外的 CNVs。与 CoDE-seq 和染色体 DNA 微阵列相比,WES 分别未能检测到 37%和 14%的 CNVs。在 82 名患者中,超过 30%的患者实现了可能的分子诊断。一半的遗传诊断由 CNVs 解释,另一半由突变解释。
CoDE-seq 已被证明具有成本效益和高效性,因为它避免了当前在临床实践中用于准确检测 CNVs 和点突变的顺序遗传筛选方法。