Hong Celine S, Singh Larry N, Mullikin James C, Biesecker Leslie G
Medical Genomics and Metabolic Genetics Branch, National Human Genome Research Institute, National Institutes of Health, Bethesda, MD, 20892, USA.
NIH Intramural Sequencing Center, National Human Genome Research Institute, National Institutes of Health, Bethesda, MD, 20852, USA.
Genome Med. 2016 Aug 8;8(1):82. doi: 10.1186/s13073-016-0336-6.
Reproducibility is receiving increased attention across many domains of science and genomics is no exception. Efforts to identify copy number variations (CNVs) from exome sequence (ES) data have been increasing. Many algorithms have been published to discover CNVs from exomes and a major challenge is the reproducibility in other datasets. Here we test exome CNV calling reproducibility under three conditions: data generated by different sequencing centers; varying sample sizes; and varying capture methodology.
Four CNV tools were tested: eXome Hidden Markov Model (XHMM), Copy Number Inference From Exome Reads (CoNIFER), EXCAVATOR, and Copy Number Analysis for Targeted Resequencing (CONTRA). To examine the reproducibility, we ran the callers on four datasets, varying sample sizes of N = 10, 30, 75, 100, 300, and data with different capture methodology. We examined the false negative (FN) calls and false positive (FP) calls for potential limitations of the CNV callers. The positive predictive value (PPV) was measured by checking the CNV call concordance against single nucleotide polymorphism array.
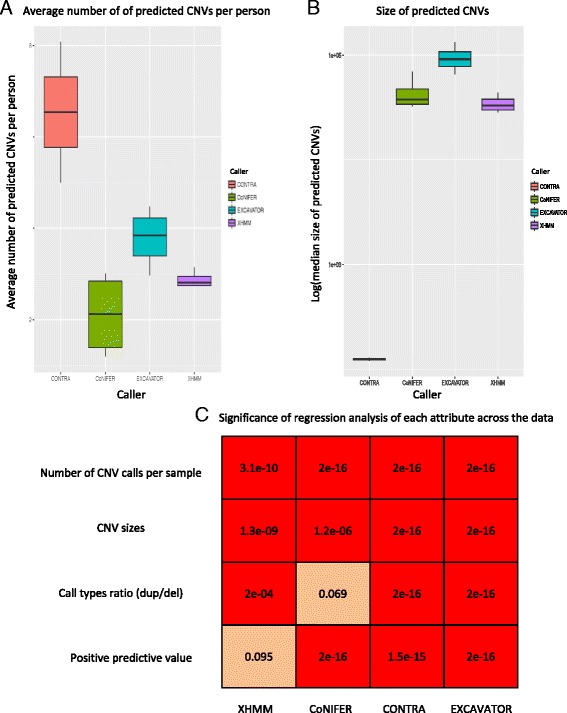
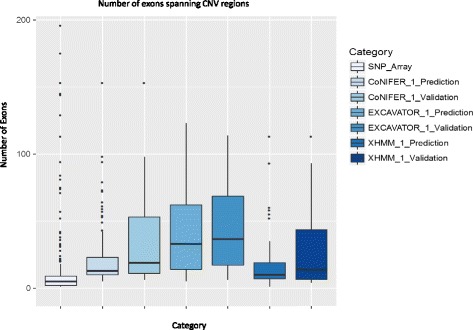
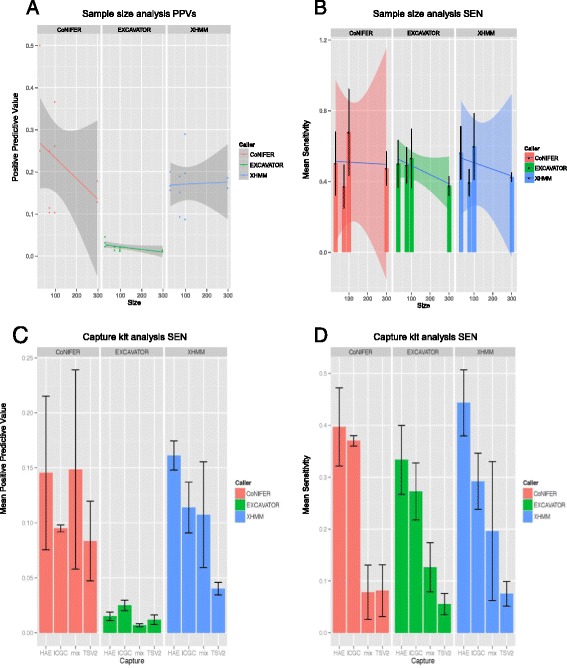
Using independently generated datasets, we examined the PPV for each dataset and observed wide range of PPVs. The PPV values were highly data dependent (p <0.001). For the sample sizes and capture method analyses, we tested the callers in triplicates. Both analyses resulted in wide ranges of PPVs, even for the same test. Interestingly, negative correlations between the PPV and the sample sizes were observed for CoNIFER (ρ = -0.80). Further examination of FN calls showed that 44 % of these were missed by all callers and were attributed to the CNV size (46 % spanned ≤3 exons). Overlap of the FP calls showed that FPs were unique to each caller, indicative of algorithm dependency.
Our results demonstrate that further improvements in CNV callers are necessary to improve reproducibility and to include wider spectrum of CNVs (including the small CNVs). These CNV callers should be evaluated on multiple independent, heterogeneously generated datasets of varying size to increase robustness and utility. These approaches to the evaluation of exome CNV are essential to support wide utility and applicability of CNV discovery in exome studies.
可重复性在许多科学领域正受到越来越多的关注,基因组学也不例外。从外显子组序列(ES)数据中识别拷贝数变异(CNV)的研究越来越多。已经发表了许多用于从外显子组中发现CNV的算法,而一个主要挑战是在其他数据集中的可重复性。在此,我们在三种条件下测试外显子组CNV检测的可重复性:不同测序中心生成的数据;不同的样本量;以及不同的捕获方法。
测试了四种CNV工具:外显子组隐马尔可夫模型(XHMM)、从外显子组读数推断拷贝数(CoNIFER)、挖掘器(EXCAVATOR)和靶向重测序的拷贝数分析(CONTRA)。为了检验可重复性,我们在四个数据集上运行这些检测工具,样本量分别为N = 10、30、75、100、300,以及采用不同捕获方法的数据。我们检查了假阴性(FN)检测和假阳性(FP)检测,以了解CNV检测工具的潜在局限性。通过将CNV检测结果与单核苷酸多态性阵列进行比对来测量阳性预测值(PPV)。
使用独立生成的数据集,我们检查了每个数据集的PPV,并观察到PPV的范围很广。PPV值高度依赖于数据(p <0.001)。对于样本量和捕获方法分析,我们对检测工具进行了三次重复测试。即使对于相同的测试,这两种分析都导致了PPV的广泛范围。有趣的是,对于CoNIFER,观察到PPV与样本量之间存在负相关(ρ = -0.80)。对FN检测的进一步检查表明,所有检测工具都遗漏了其中百分之44的检测结果,这归因于CNV的大小(百分之46跨越≤3个外显子)。FP检测的重叠显示,FP对于每个检测工具都是独特的,这表明算法依赖性。
我们的结果表明,需要进一步改进CNV检测工具,以提高可重复性并纳入更广泛的CNV谱(包括小CNV)。这些CNV检测工具应在多个独立的、异质生成的不同大小数据集上进行评估,以提高稳健性和实用性。这些评估外显子组CNV的方法对于支持CNV发现在外显子组研究中的广泛应用和适用性至关重要。