Centre for Respiratory Diseases and Meningitis, National Institute for Communicable Diseases of the National Health Laboratory Service, Johannesburg, South Africa.
Division of Medical Virology, Department of Pathology, University of Cape Town, Cape Town, South Africa.
PLoS One. 2018 May 24;13(5):e0198101. doi: 10.1371/journal.pone.0198101. eCollection 2018.
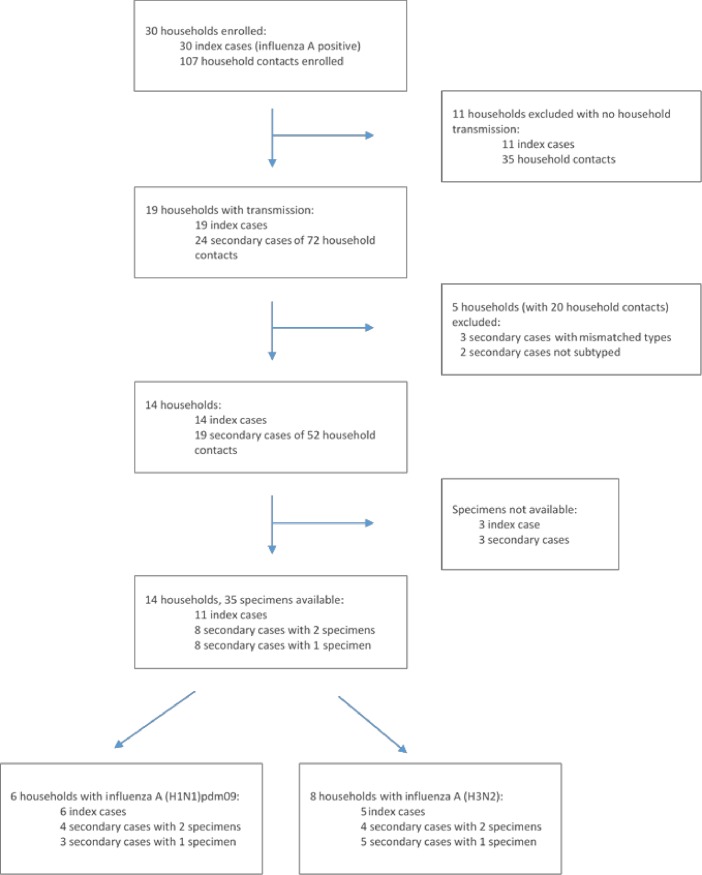
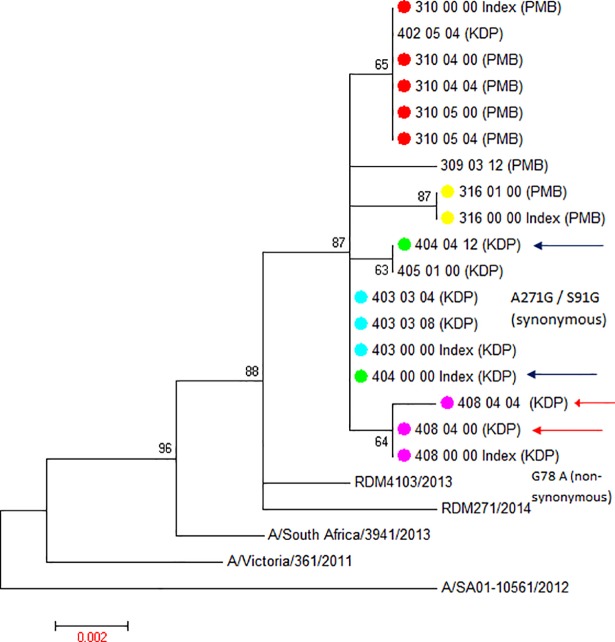
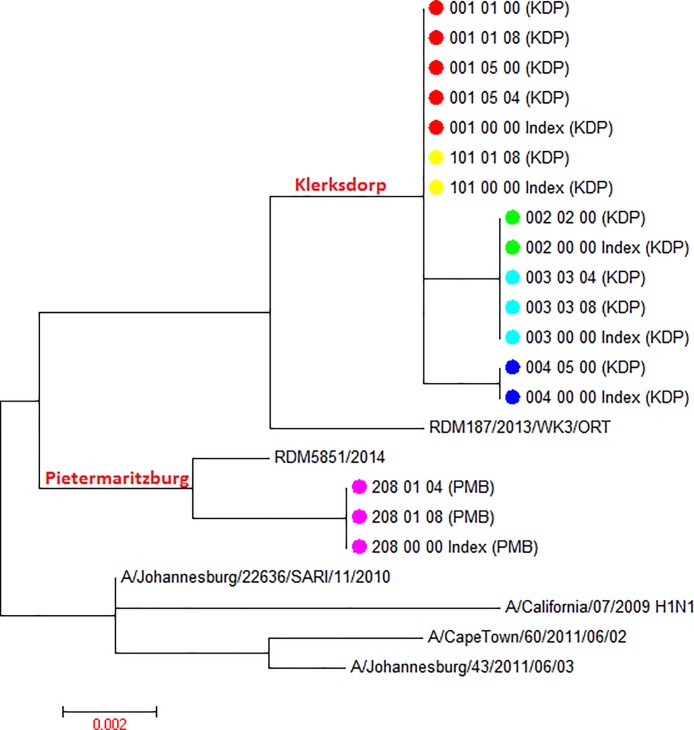
Limited information is available on influenza virus sequence drift between transmission events. In countries with high HIV burdens, like South Africa, the direct and indirect effect of HIV on influenza sequence drift between transmission events may be of public health concern. To this end, we measured hemagglutinin sequence diversity between influenza transmission events using data and specimens from a study investigating household transmission dynamics of seasonal influenza viruses in 2 peri-urban communities in South Africa during the 2013 influenza season. Thirty index cases and 107 of 110 eligible household contacts were enrolled into the study, 47% (14/30) demonstrating intra-household laboratory-confirmed influenza transmission. In this study 35 partial hemagglutinin gene sequences were obtained by Sanger sequencing from 11 index cases (sampled at enrolment only) and 16 secondary cases (8 cases sampled at 1 and 8 cases sampled at 2 time-points). Viral sequence identities confirmed matched influenza transmission pairs within the 11 households with corresponding sequenced index and secondary cases. Phylogenetic analysis revealed 10 different influenza viral lineages in the 14 households. Influenza A(H1N1)pdm09 strains were shown to be genetically distinct between the 2 communities (from distinct geographic regions), which was not observed for the influenza A(H3N2) strains. Intra-host/intra-household influenza A(H3N2) sequence drift was identified in 2 households. The first was a synonymous mutation between the index case and a household contact, and the second a non-synonymous mutation between 2 serial samples taken at days 0 and 4 post enrolment from an HIV-infected secondary case. Limited inter-household sequence diversity was observed as highlighted by sharing of the same influenza strain between different households within each community. The limited intra-household sequence drift is in line with previous studies also using Sanger sequencing, corroborating the presence of strict selective bottlenecks that limit sequence variance. We were not able to directly ascertain the effect of HIV on influenza sequence drift between transmission events.
关于传播事件之间流感病毒序列漂移的信息有限。在像南非这样艾滋病毒负担高的国家,艾滋病毒对传播事件之间流感序列漂移的直接和间接影响可能引起公共卫生关注。为此,我们使用来自南非两个城市社区季节性流感病毒家庭传播动力学研究的数据和标本,测量了传播事件之间流感血凝素序列的多样性。在这项研究中,30 名索引病例和 110 名符合条件的家庭接触者中的 107 名被纳入研究,47%(14/30)显示家庭内实验室确诊的流感传播。在这项研究中,从 11 名索引病例(仅在入组时采样)和 16 名二级病例(8 名在 1 时间点采样,8 名在 2 时间点采样)中通过 Sanger 测序获得了 35 个部分血凝素基因序列。病毒序列同一性证实了在 11 户家庭中与相应测序的索引和二级病例相匹配的流感传播对。系统发育分析显示,在 14 户家庭中存在 10 种不同的流感病毒谱系。研究表明,来自两个不同地理区域的两个社区的甲型 H1N1pdm09 株在遗传上是不同的,而甲型 H3N2 株则不是。在 2 个家庭中发现了宿主/家庭内的甲型 H3N2 序列漂移。第一个是在索引病例和一个家庭接触者之间的同义突变,第二个是在感染艾滋病毒的二级病例入组后第 0 和 4 天采集的两个连续样本之间的非同义突变。由于在每个社区中不同家庭之间共享相同的流感株,因此观察到有限的家庭间序列多样性。有限的家庭内序列漂移与之前也使用 Sanger 测序的研究一致,证实了严格的选择瓶颈的存在,限制了序列变异。我们无法直接确定艾滋病毒对传播事件之间流感序列漂移的影响。