Heit Yonaton N, Nanda Kaushik D, Beran Gregory J O
Department of Chemistry , University of California , Riverside , California 92521 , USA . Email:
Chem Sci. 2016 Jan 1;7(1):246-255. doi: 10.1039/c5sc03014e. Epub 2015 Sep 29.
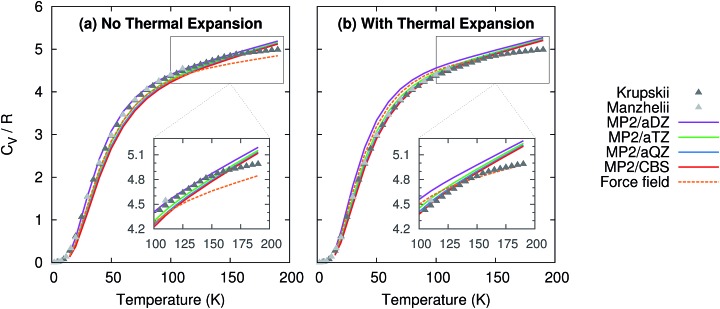
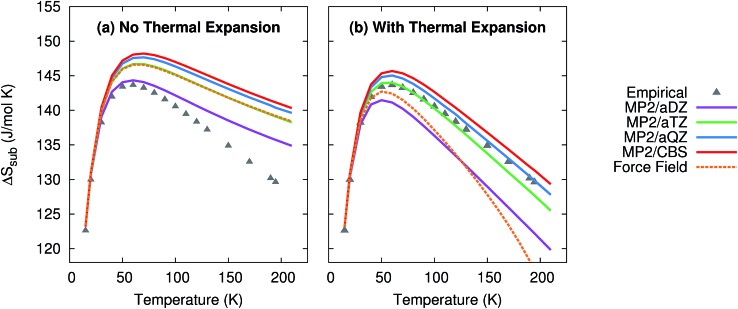
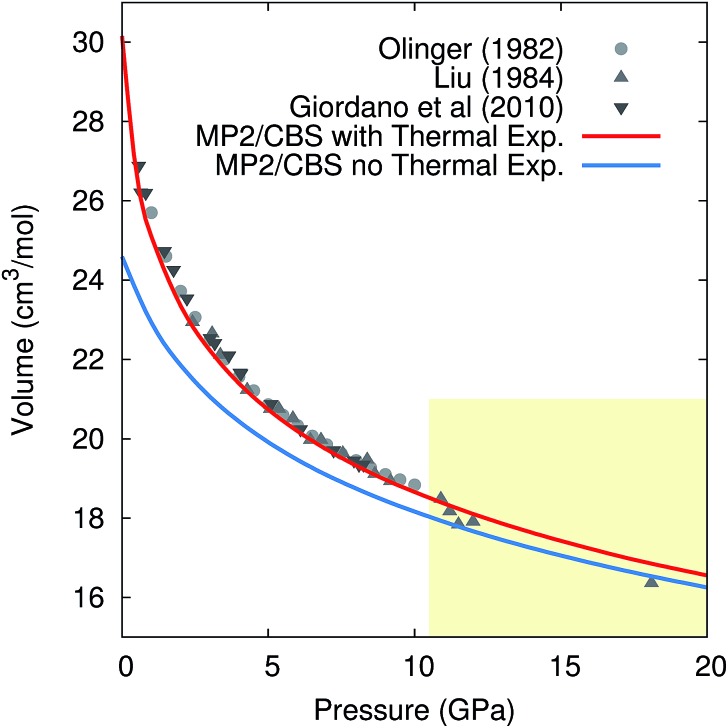
Molecular crystal structures, thermodynamics, and mechanical properties can vary substantially with temperature, and predicting these temperature-dependencies correctly is important for many practical applications in the pharmaceutical industry and other fields. However, most electronic structure predictions of molecular crystal properties neglect temperature and/or thermal expansion, leading to potentially erroneous results. Here, we demonstrate that by combining large basis set second-order Møller-Plesset (MP2) or even coupled cluster singles, doubles, and perturbative triples (CCSD(T)) electronic structure calculations with a quasiharmonic treatment of thermal expansion, experimentally observable properties such as the unit cell volume, heat capacity, enthalpy, entropy, sublimation point and bulk modulus of phase I crystalline carbon dioxide can be predicted in excellent agreement with experiment over a broad range of temperatures. These results point toward a promising future for prediction of molecular crystal properties at real-world temperatures and pressures.
分子晶体结构、热力学和力学性能会随温度发生显著变化,正确预测这些温度依赖性对于制药行业和其他领域的许多实际应用至关重要。然而,大多数分子晶体性质的电子结构预测都忽略了温度和/或热膨胀,从而可能导致错误的结果。在此,我们证明,通过将大基组二阶莫勒-普列斯勒(MP2)甚至耦合簇单双激发和微扰三激发(CCSD(T))电子结构计算与热膨胀的准谐处理相结合,可以在很宽的温度范围内预测出与实验结果高度吻合的实验可观测性质,如晶胞体积、热容、焓、熵、升华点和I相结晶二氧化碳的体积模量。这些结果表明,在实际温度和压力下预测分子晶体性质有着光明的前景。