Greenwell Chandler, McKinley Jessica L, Zhang Peiyu, Zeng Qun, Sun Guangxu, Li Bochen, Wen Shuhao, Beran Gregory J O
Department of Chemistry , University of California , Riverside , California 92521 , USA . Email:
Xtalpi, Inc. , 245 Main St, 12th Floor , Cambridge , MA 02142 , USA.
Chem Sci. 2020 Jan 14;11(8):2200-2214. doi: 10.1039/c9sc05689k. eCollection 2020 Feb 28.
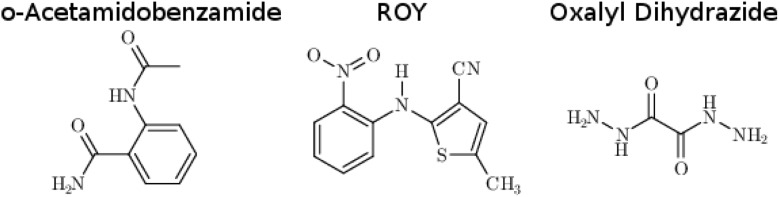
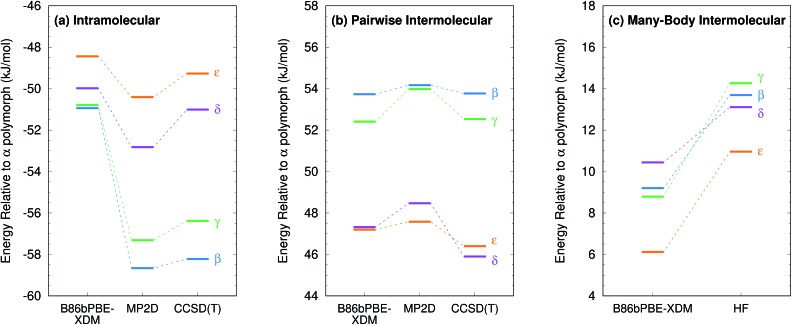
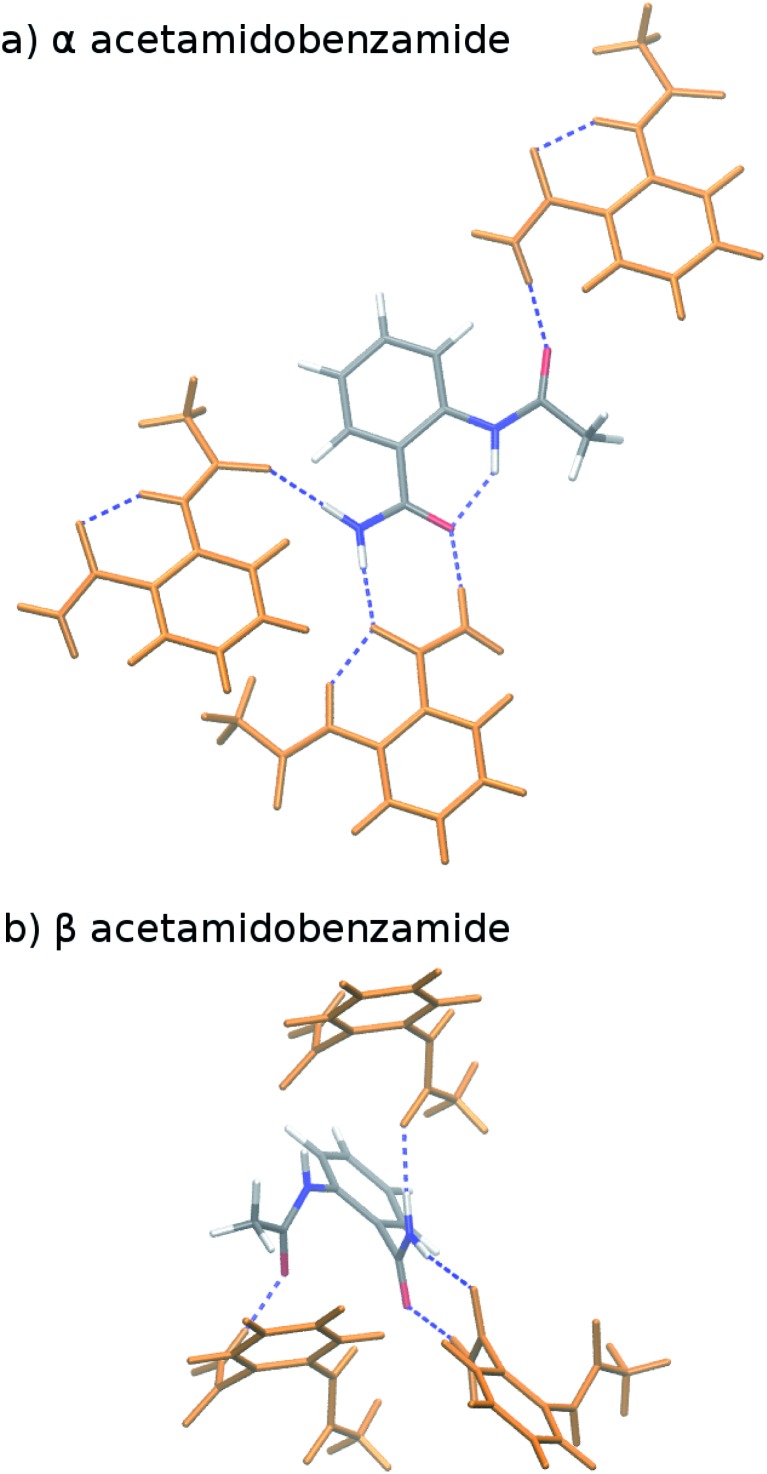
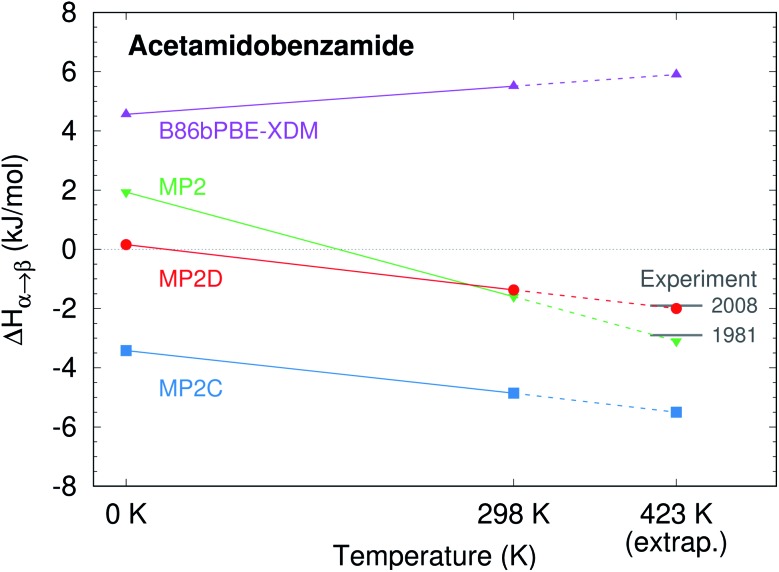
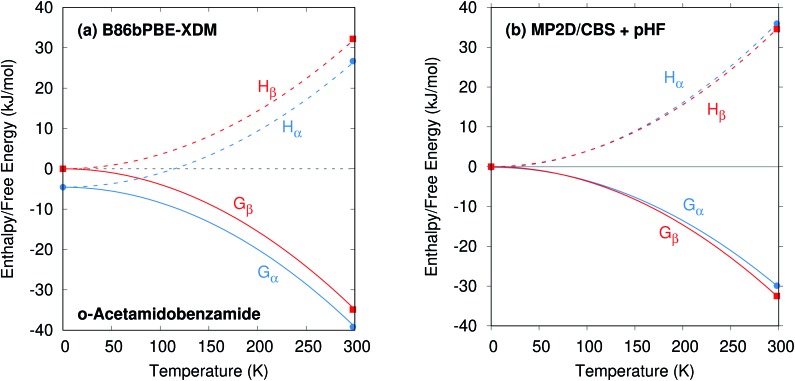
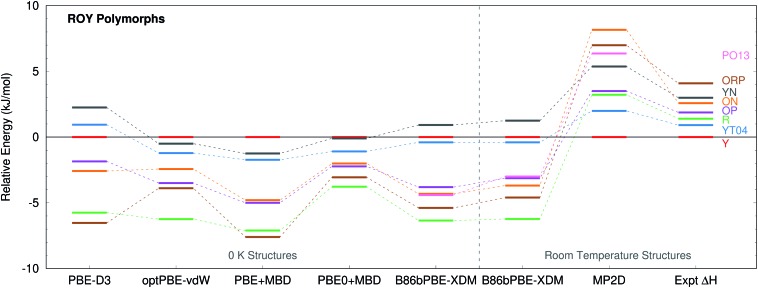
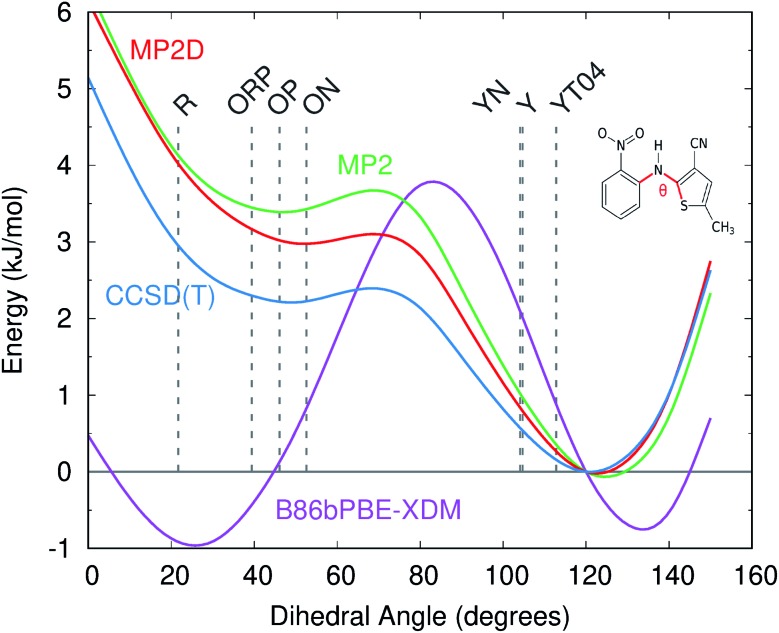
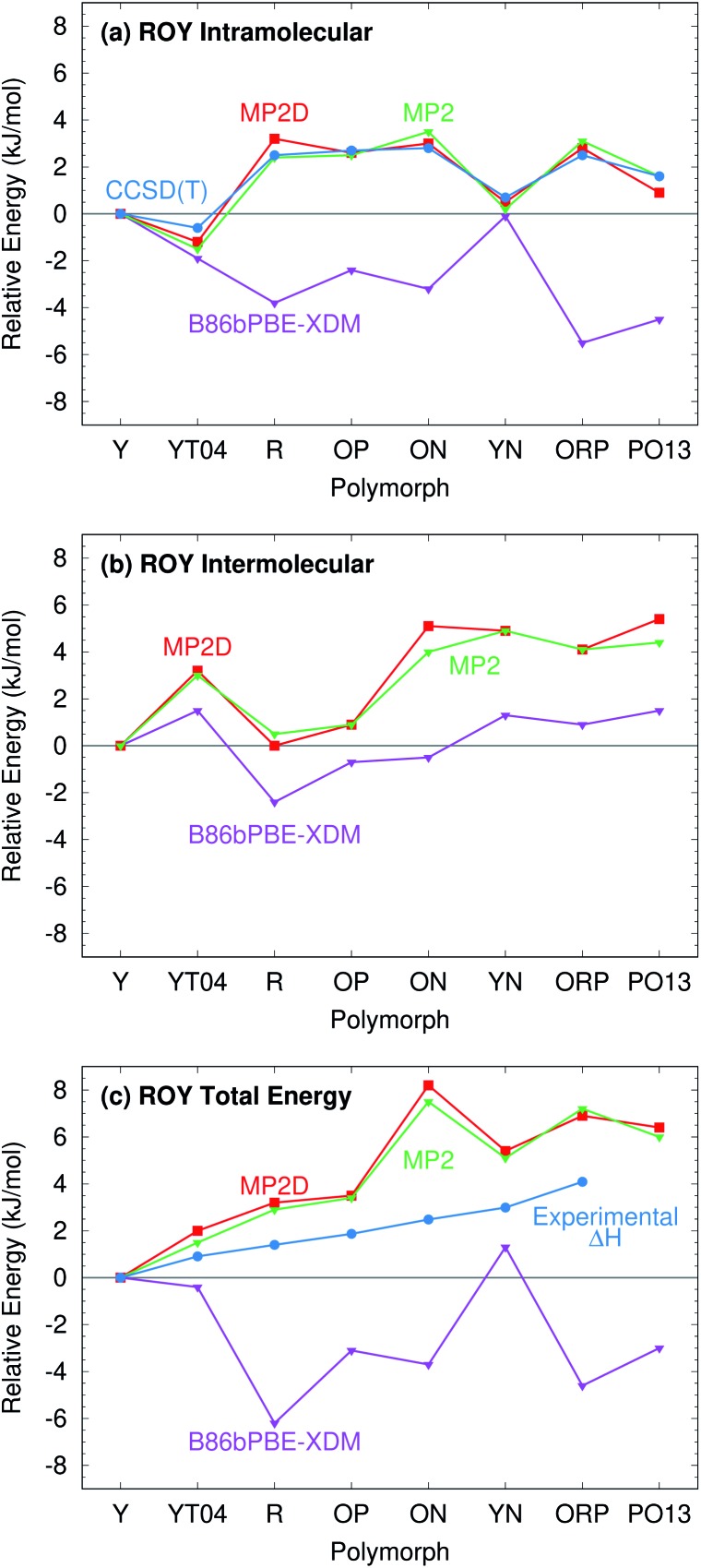
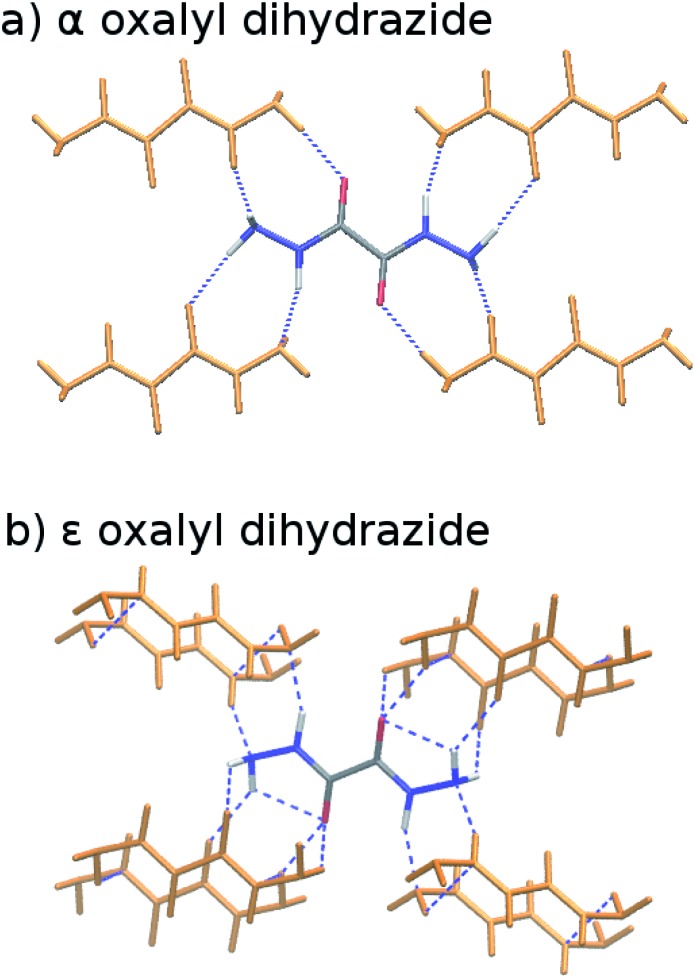
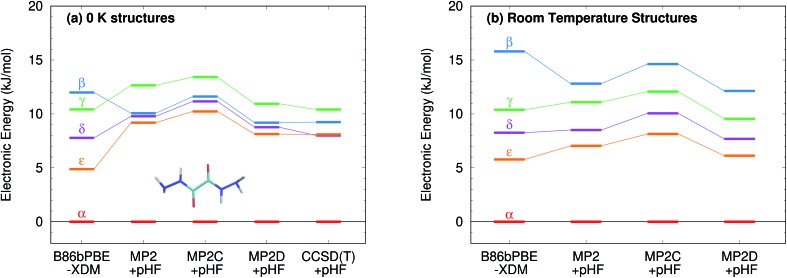
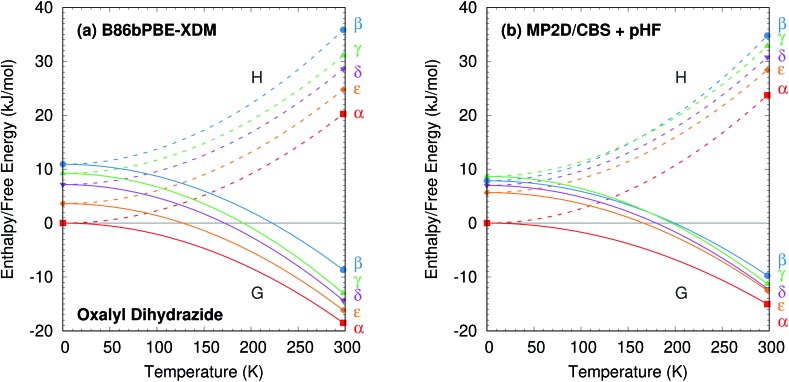
Molecular crystal structure prediction is increasingly being applied to study the solid form landscapes of larger, more flexible pharmaceutical molecules. Despite many successes in crystal structure prediction, van der Waals-inclusive density functional theory (DFT) methods exhibit serious failures predicting the polymorph stabilities for a number of systems exhibiting conformational polymorphism, where changes in intramolecular conformation lead to different intermolecular crystal packings. Here, the stabilities of the conformational polymorphs of -acetamidobenzamide, ROY, and oxalyl dihydrazide are examined in detail. DFT functionals that have previously been very successful in crystal structure prediction perform poorly in all three systems, due primarily to the poor intramolecular conformational energies, but also due to the intermolecular description in oxalyl dihydrazide. In all three cases, a fragment-based dispersion-corrected second-order Møller-Plesset perturbation theory (MP2D) treatment of the crystals overcomes these difficulties and predicts conformational polymorph stabilities in good agreement with experiment. These results highlight the need for methods which go beyond current-generation DFT functionals to make crystal polymorph stability predictions truly reliable.
分子晶体结构预测正越来越多地应用于研究更大、更具柔性的药物分子的固态形式格局。尽管在晶体结构预测方面取得了许多成功,但包含范德华力的密度泛函理论(DFT)方法在预测一些呈现构象多晶型现象的体系的多晶型稳定性时表现出严重失败,在这些体系中分子内构象的变化会导致不同的分子间晶体堆积。在此,对乙酰氨基苯甲酰胺、ROY和草酰二肼的构象多晶型的稳定性进行了详细研究。此前在晶体结构预测中非常成功的DFT泛函在这三个体系中均表现不佳,主要原因是分子内构象能量较差,同时也是由于草酰二肼中的分子间描述问题。在所有这三种情况下,基于片段的色散校正二阶莫勒 - 普列斯特定则微扰理论(MP2D)对晶体的处理克服了这些困难,并预测出与实验结果高度一致的构象多晶型稳定性。这些结果凸显了需要超越当前一代DFT泛函的方法,以使晶体多晶型稳定性预测真正可靠。