Otto Thomas D, Böhme Ulrike, Sanders Mandy, Reid Adam, Bruske Ellen I, Duffy Craig W, Bull Pete C, Pearson Richard D, Abdi Abdirahman, Dimonte Sandra, Stewart Lindsay B, Campino Susana, Kekre Mihir, Hamilton William L, Claessens Antoine, Volkman Sarah K, Ndiaye Daouda, Amambua-Ngwa Alfred, Diakite Mahamadou, Fairhurst Rick M, Conway David J, Franck Matthias, Newbold Chris I, Berriman Matt
Wellcome Sanger Institute, Hinxton, UK.
Centre of Immunobiology, Institute of Infection, Immunity & Inflammation, College of Medical, Veterinary and Life Sciences, University of Glasgow, Glasgow, UK.
Wellcome Open Res. 2018 May 3;3:52. doi: 10.12688/wellcomeopenres.14571.1. eCollection 2018.
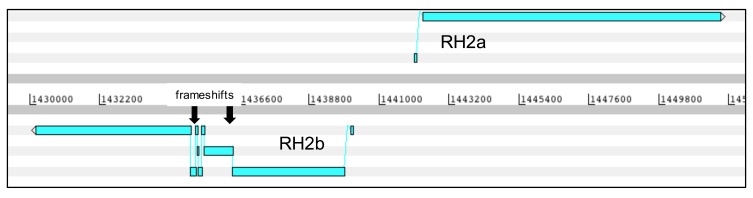
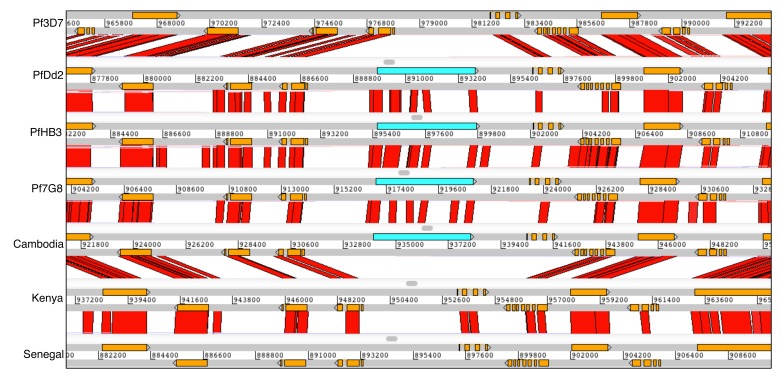
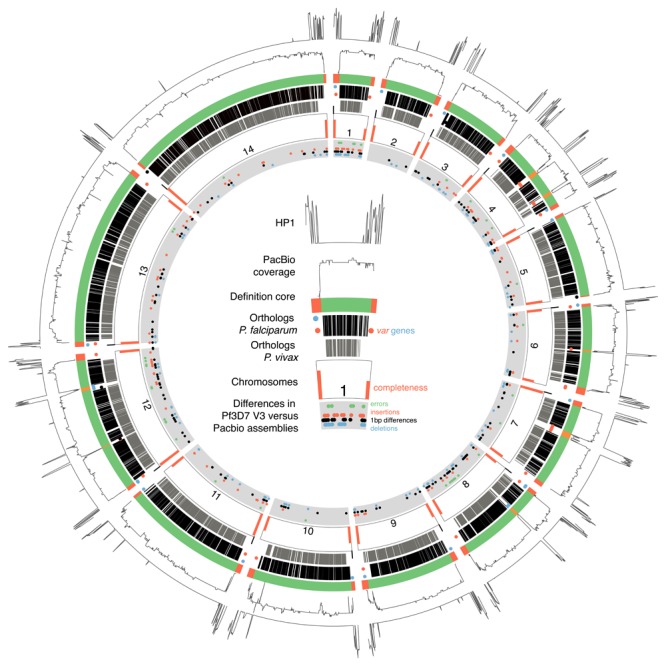
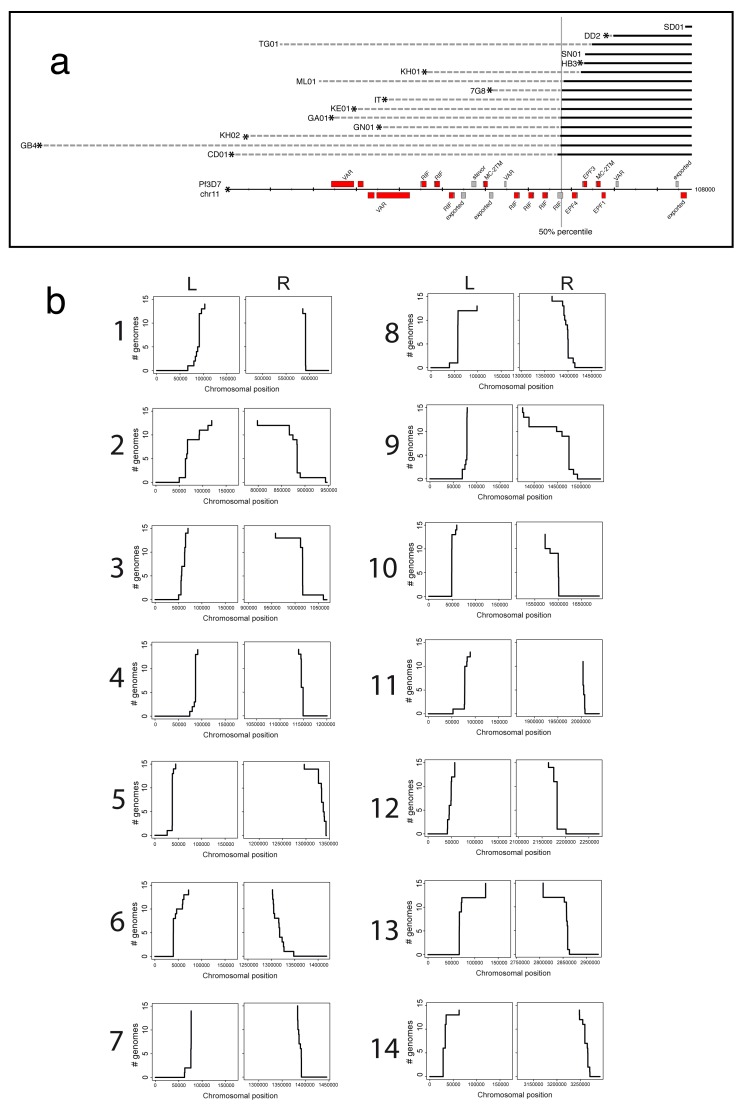
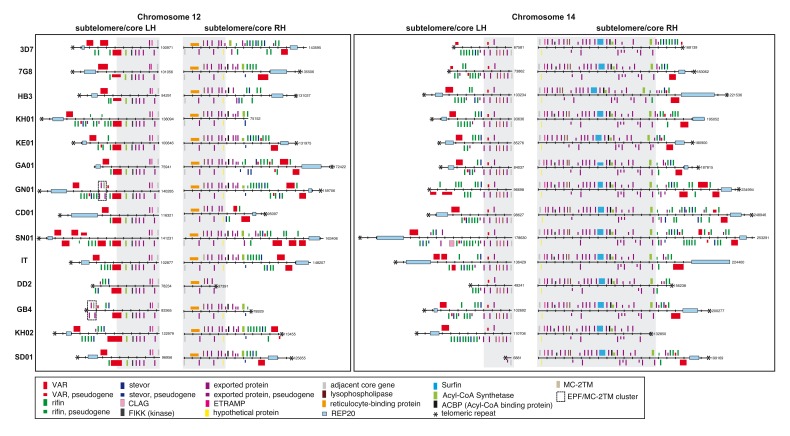
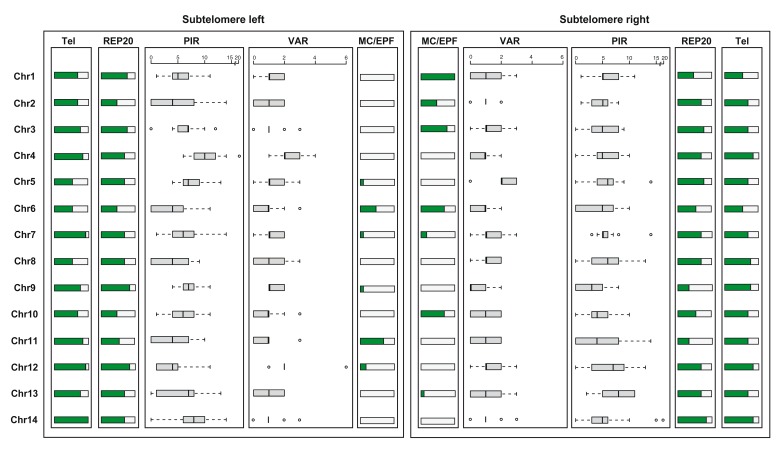
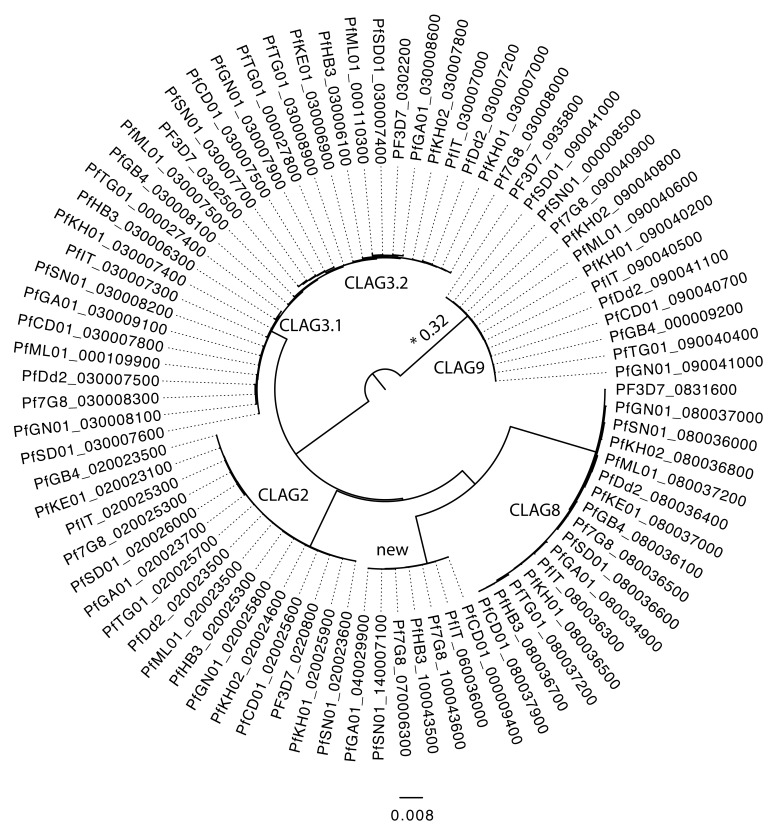
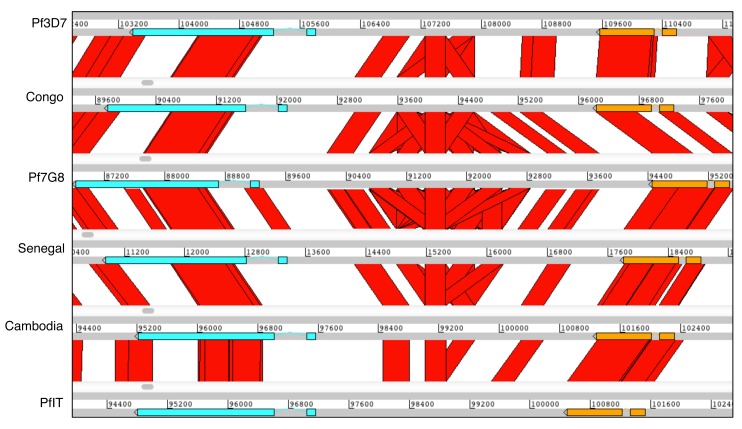
: Although thousands of clinical isolates of are being sequenced and analysed by short read technology, the data do not resolve the highly variable subtelomeric regions of the genomes that contain polymorphic gene families involved in immune evasion and pathogenesis. There is also no current standard definition of the boundaries of these variable subtelomeric regions. : Using long-read sequence data (Pacific Biosciences SMRT technology), we assembled and annotated the genomes of 15 isolates, ten of which are newly cultured clinical isolates. We performed comparative analysis of the entire genome with particular emphasis on the subtelomeric regions and the internal genes clusters. : The nearly complete sequence of these 15 isolates has enabled us to define a highly conserved core genome, to delineate the boundaries of the subtelomeric regions, and to compare these across isolates. We found highly structured variable regions in the genome. Some exported gene families purportedly involved in release of merozoites show copy number variation. As an example of ongoing genome evolution, we found a novel CLAG gene in six isolates. We also found a novel gene that was relatively enriched in the South East Asian isolates compared to those from Africa. : These 15 manually curated new reference genome sequences with their nearly complete subtelomeric regions and fully assembled genes are an important new resource for the malaria research community. We report the overall conserved structure and pattern of important gene families and the more clearly defined subtelomeric regions.
尽管数以千计的临床分离株正在通过短读长技术进行测序和分析,但这些数据无法解析基因组中高度可变的亚端粒区域,这些区域包含参与免疫逃避和发病机制的多态基因家族。目前也没有关于这些可变亚端粒区域边界的标准定义。:利用长读长序列数据(太平洋生物科学公司的单分子实时测序技术),我们组装并注释了15个疟原虫分离株的基因组,其中10个是新培养的临床分离株。我们对整个基因组进行了比较分析,特别关注亚端粒区域和内部疟原虫基因簇。:这15个分离株的近乎完整的序列使我们能够定义一个高度保守的核心基因组,划定亚端粒区域的边界,并在不同分离株之间进行比较。我们在基因组中发现了高度结构化的可变区域。一些据称参与裂殖子释放的输出基因家族显示出拷贝数变异。作为基因组持续进化的一个例子,我们在6个分离株中发现了一个新的CLAG基因。我们还发现了一个新基因,与来自非洲的分离株相比,它在东南亚分离株中相对富集。:这15个经过人工精心整理的新参考基因组序列及其近乎完整的亚端粒区域和完全组装的基因,是疟疾研究界的重要新资源。我们报告了重要基因家族的整体保守结构和模式以及定义更清晰的亚端粒区域。