Big Data Decision Institute, Jinan University, Tianhe, Guangzhou, P.R. China.
Sci Rep. 2018 Jun 11;8(1):8906. doi: 10.1038/s41598-018-27298-0.
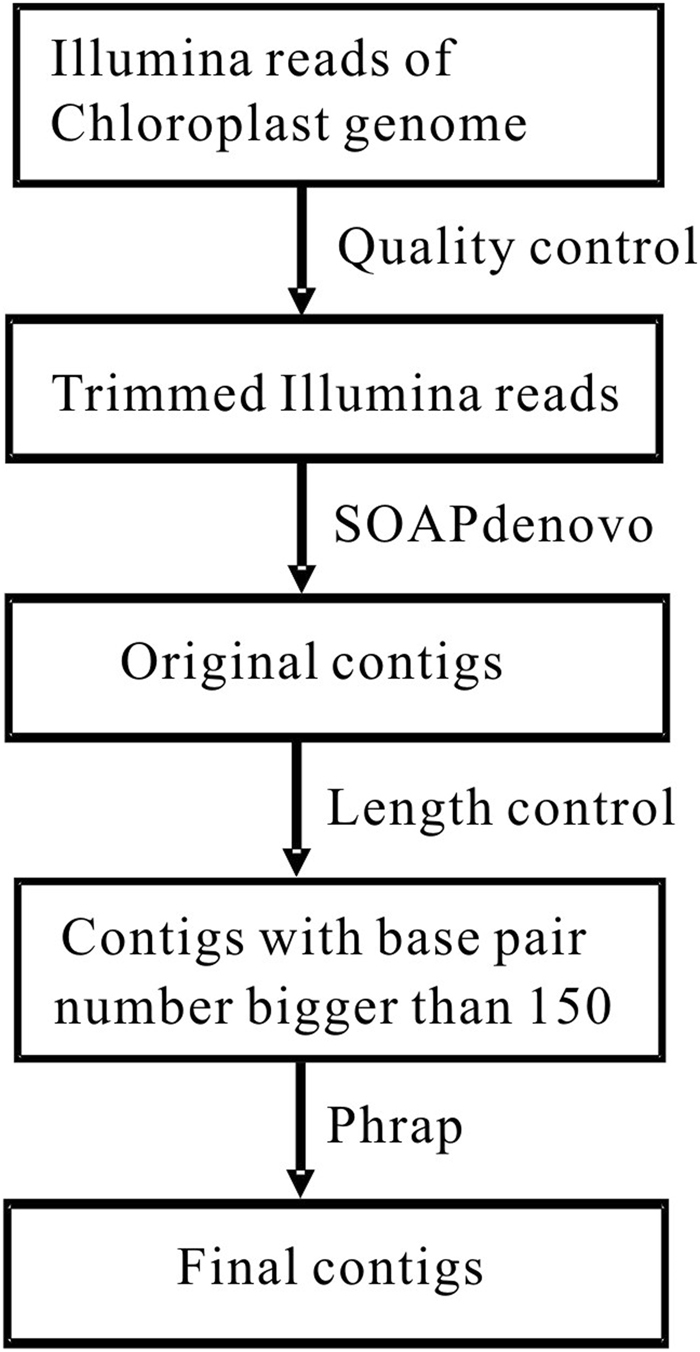
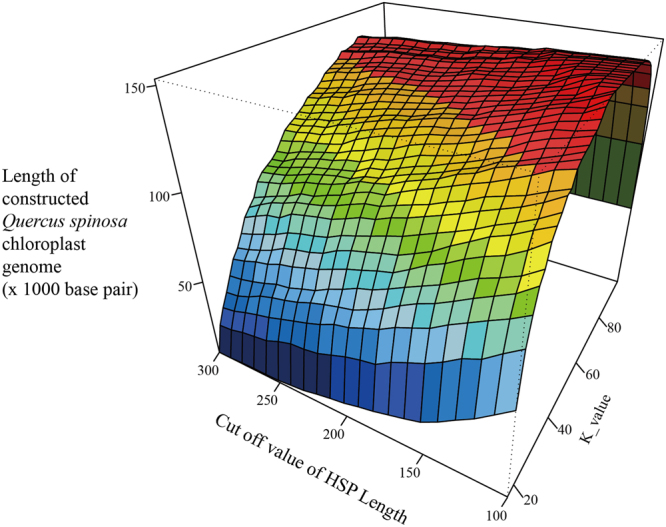
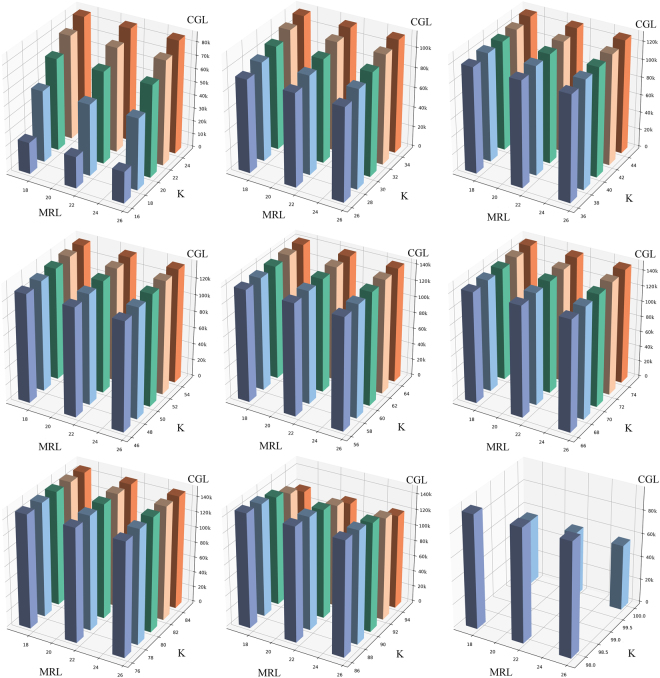
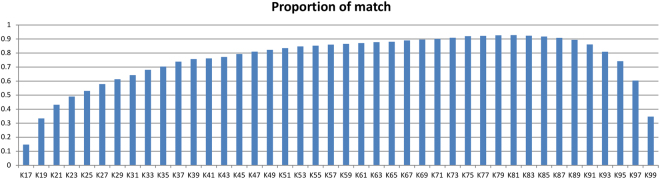
Obtaining chloroplast (cp) genome sequence is necessary for studying physiological roles in plants. However, it is difficult to use traditional sequencing methods to get cp genome sequences because of the complex procedures of preparing templates. With the advent of next-generation sequencing technology, massive genome sequences can be produced. Thus, a good pipeline to assemble next-generation sequence reads with optimized k-mer length is essential to get whole cp genome sequences. Moreover, adjustment of other parameters is also very important, especially for the assembly of the cp genome. In this study, we developed a pipeline to generate the cp genome for Quercus spinosa. When Quercus rubra was used as a reference, we achieved coverage of 97.75% after optimizing k-mer length as well as other parameters. The efficiency of the pipeline makes it a useful method for cp genome construction in plants. It also provides great perspective on the analysis of cp genome characteristics and evolution.
获得叶绿体(cp)基因组序列对于研究植物的生理作用是必要的。然而,由于模板制备过程复杂,使用传统测序方法很难获得 cp 基因组序列。随着新一代测序技术的出现,可以产生大量的基因组序列。因此,采用优化的 k-mer 长度来组装下一代序列读取的良好流水线对于获得完整的 cp 基因组序列是必不可少的。此外,其他参数的调整也非常重要,特别是对于 cp 基因组的组装。在这项研究中,我们开发了一个生成 Quercus spinosa cp 基因组的流水线。当使用 Quercus rubra 作为参考时,我们通过优化 k-mer 长度以及其他参数,实现了 97.75%的覆盖率。该流水线的效率使其成为植物 cp 基因组构建的一种有用方法。它还为 cp 基因组特征和进化的分析提供了广阔的视角。