Institute of Forest Biotechnology, Forestry College, Hebei Agricultural University, Baoding, 071000, China.
Hebei Key Laboratory for Tree Genetic Resources and Forest Protection, Baoding, 071000, China.
Sci Rep. 2022 Sep 24;12(1):15953. doi: 10.1038/s41598-022-20184-w.
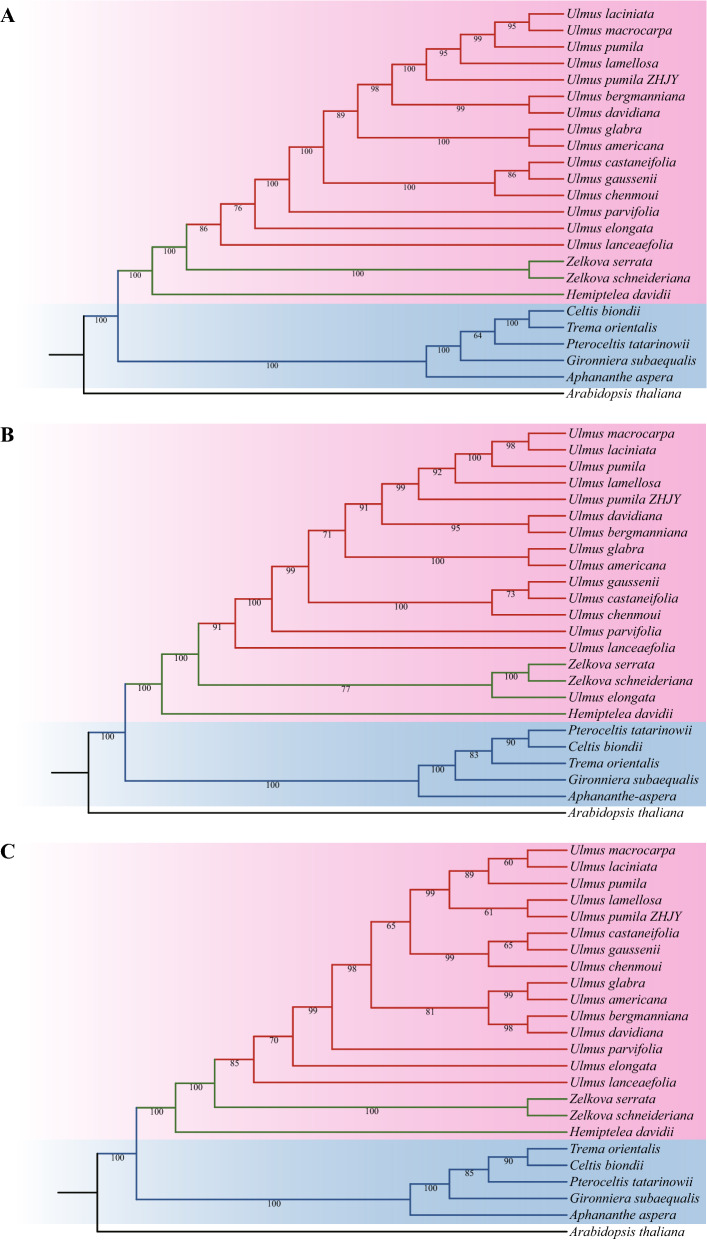
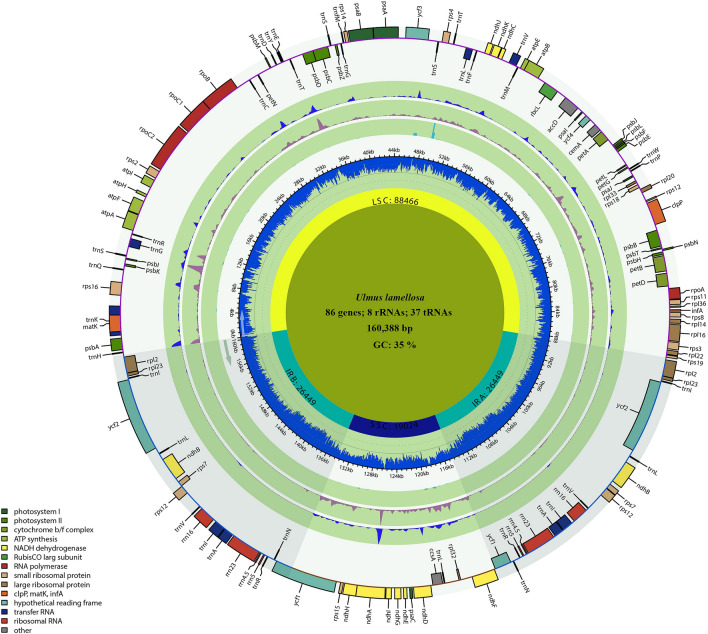
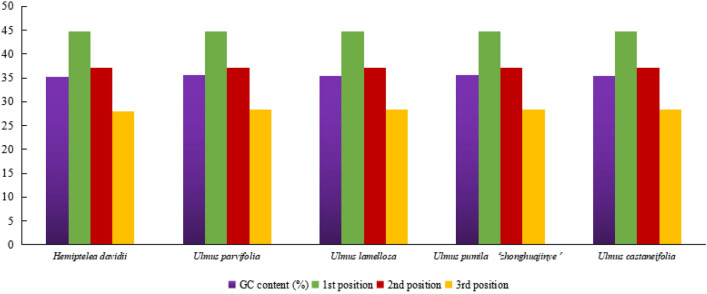
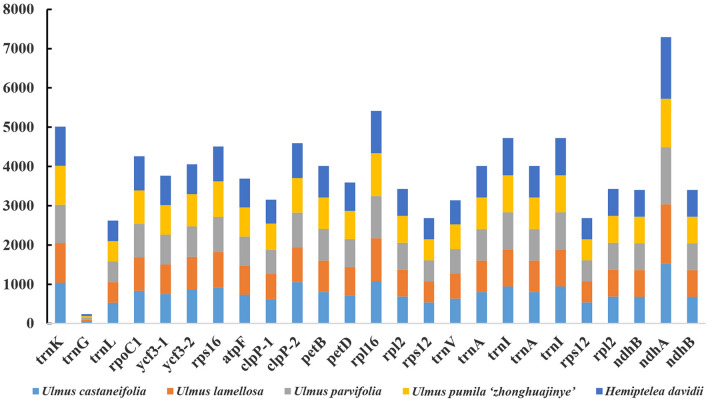
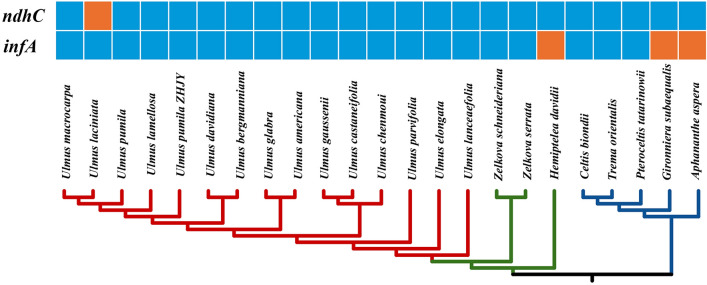
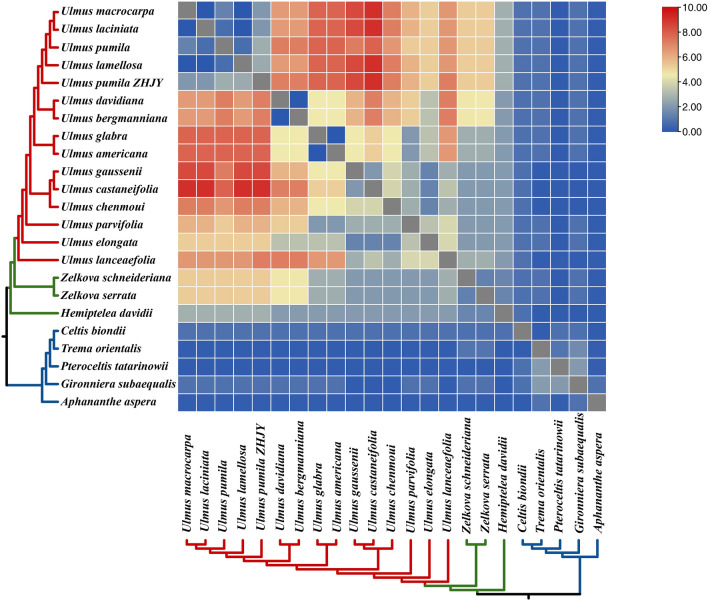
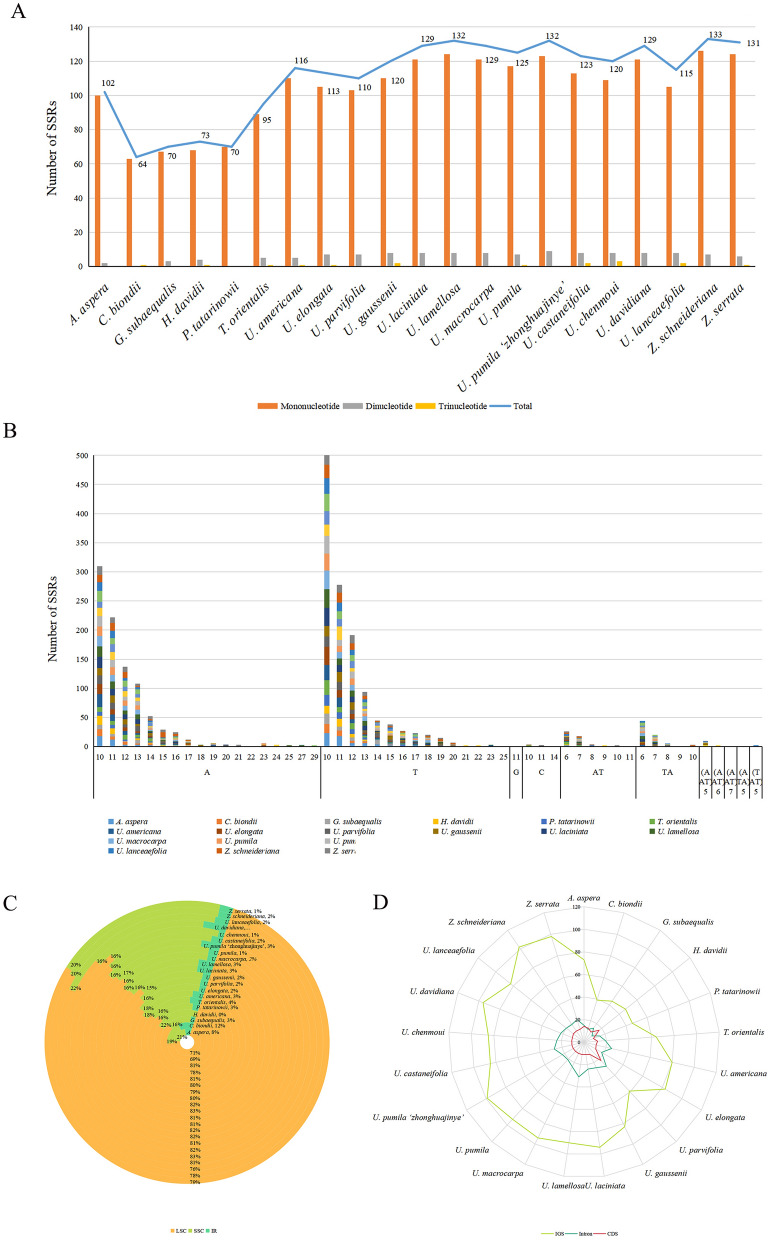
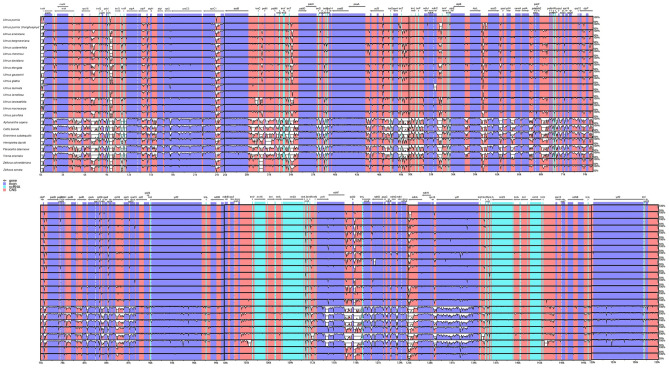
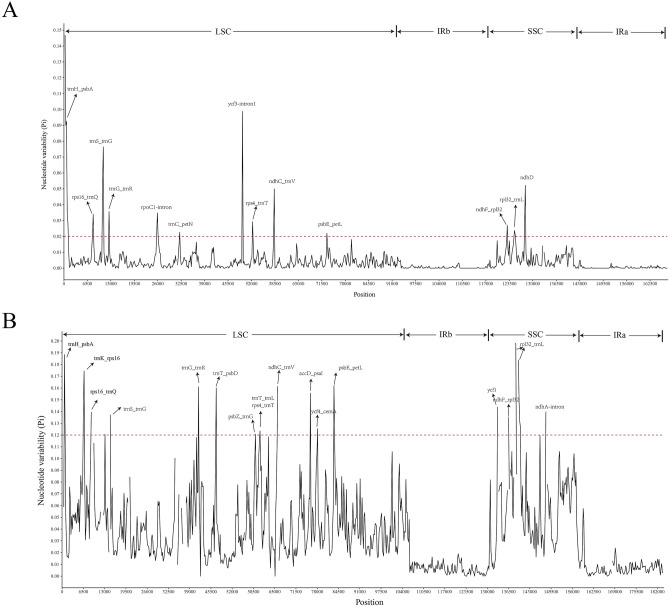
In this study, the chloroplast (cp) genomes of Hemiptelea davidii, Ulmus parvifolia, Ulmus lamellosa, Ulmus castaneifolia, and Ulmus pumila 'zhonghuajinye' were spliced, assembled and annotated using the Illumina HiSeq PE150 sequencing platform, and then compared to the cp genomes of other Ulmus and Ulmaceae species. The results indicated that the cp genomes of the five sequenced species showed a typical tetrad structure with full lengths ranging from 159,113 to 160,388 bp. The large single copy (LSC), inverted repeat (IR), and small single copy (SSC) lengths were in the range of 87,736-88,466 bp, 26,317-26,622 bp and 18,485-19,024 bp, respectively. A total of 130-131 genes were annotated, including 85-86 protein-coding genes, 37 tRNA genes and eight rRNA genes. The GC contents of the five species were similar, ranging from 35.30 to 35.62%. Besides, the GC content was different in different region and the GC content in IR region was the highest. A total of 64-133 single sequence repeat (SSR) loci were identified among all 21 Ulmaceae species. The (A) and (T) types of mononucleotide were highest in number, and the lengths were primarily distributed in 10-12 bp, with a clear AT preference. A branch-site model and a Bayes Empirical Bayes analysis indicated that the rps15 and rbcL had the positive selection sites. Besides, the analysis of mVISTA and sliding windows got a lot of hotspots such as trnH/psbA, rps16/trnQ, trnS/trnG, trnG/trnR and rpl32/trnL, which could be utilized as potential markers for the species identification and phylogeny reconstruction within Ulmus in the further studies. Moreover, the evolutionary tree of Ulmaceae species based on common protein genes, whole cp genome sequences and common genes in IR region of the 23 Ulmaceae species were constructed using the ML method. The results showed that these Ulmaceae species were divided into two branches, one that included Ulmus, Zelkova and Hemiptelea, among which Hemiptelea was the first to differentiate and one that included Celtis, Trema, Pteroceltis, Gironniera and Aphananthe. Besides, these variations found in this study could be used for the classification, identification and phylogenetic study of Ulmus species. Our study provided important genetic information to support further investigations into the phylogenetic development and adaptive evolution of Ulmus and Ulmaceae species.
在这项研究中,我们使用 Illumina HiSeq PE150 测序平台拼接、组装和注释了麻叶绣球、榆叶梅、新疆杨、毛白杨和中华金叶榆的叶绿体(cp)基因组,并与其他榆属和榆科物种的 cp 基因组进行了比较。结果表明,这 5 个测序物种的 cp 基因组呈现典型的四联体结构,全长范围为 159113-160388bp。大单拷贝(LSC)、反向重复(IR)和小单拷贝(SSC)的长度分别在 87736-88466bp、26317-26622bp和 18485-19024bp范围内。总共注释了 130-131 个基因,包括 85-86 个蛋白编码基因、37 个 tRNA 基因和 8 个 rRNA 基因。这 5 个物种的 GC 含量相似,范围在 35.30-35.62%之间。此外,不同区域的 GC 含量存在差异,IR 区域的 GC 含量最高。在 21 个榆科物种中总共鉴定出 64-133 个单核苷酸重复(SSR)位点。单核苷酸的(A)和(T)类型数量最多,长度主要分布在 10-12bp,具有明显的 AT 偏好性。分支位点模型和贝叶斯经验贝叶斯分析表明,rps15 和 rbcL 具有阳性选择位点。此外,mVISTA 和滑动窗口分析得到了大量热点,如 trnH/psbA、rps16/trnQ、trnS/trnG、trnG/trnR 和 rpl32/trnL,可作为进一步研究中榆属种间鉴定和系统发育重建的潜在标记。此外,使用 ML 方法构建了基于 23 个榆科物种的共同蛋白基因、整个 cp 基因组序列和 IR 区共同基因的榆科物种进化树。结果表明,这些榆科物种分为两个分支,一个分支包括榆属、朴属和麻叶绣球属,其中麻叶绣球属最先分化,另一个分支包括朴属、榆属、青檀属、榔榆属和榉属。此外,本研究中的这些变异可用于榆属物种的分类、鉴定和系统发育研究。本研究提供了重要的遗传信息,支持进一步研究榆属和榆科物种的系统发育发展和适应性进化。