Genetics and Molecular Biology Program, Emory University, Atlanta, GA, USA.
Department of Human Genetics, Emory University School of Medicine, Atlanta, GA, USA.
BMC Genomics. 2018 Jun 19;19(1):476. doi: 10.1186/s12864-018-4842-3.
Gene expression can be influenced by DNA methylation 1) distally, at regulatory elements such as enhancers, as well as 2) proximally, at promoters. Our current understanding of the influence of distal DNA methylation changes on gene expression patterns is incomplete. Here, we characterize genome-wide methylation and expression patterns for ~ 13 k genes to explore how DNA methylation interacts with gene expression, throughout the genome.
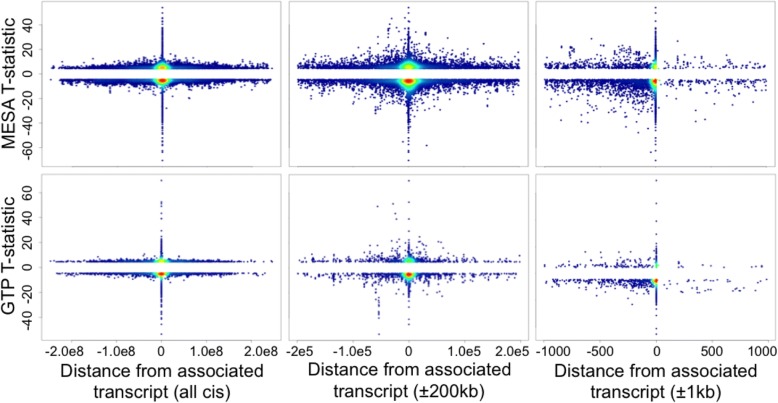
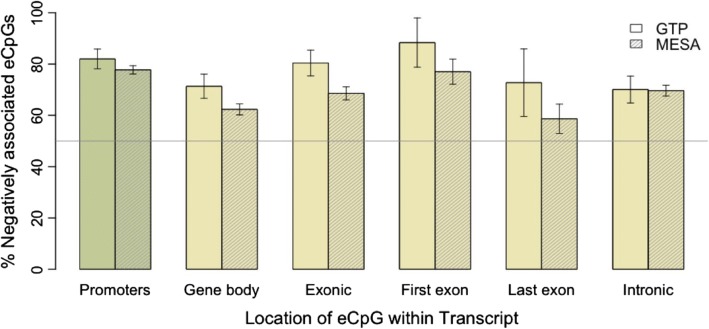
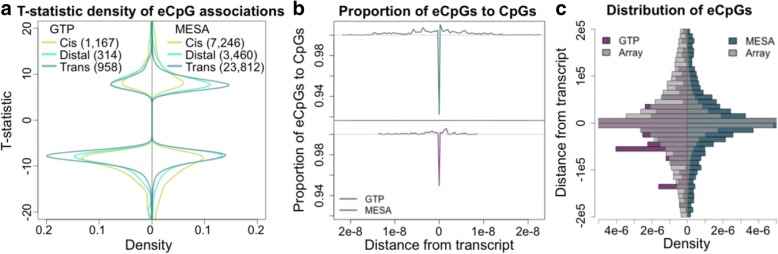
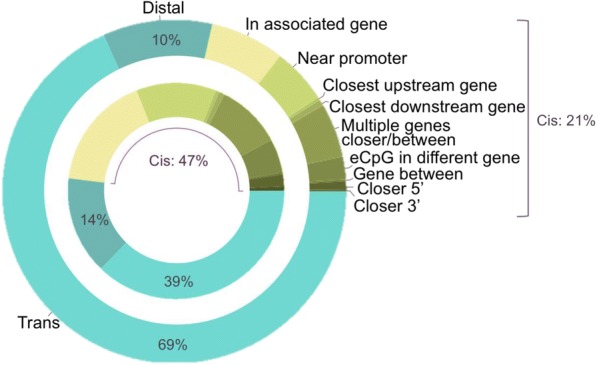
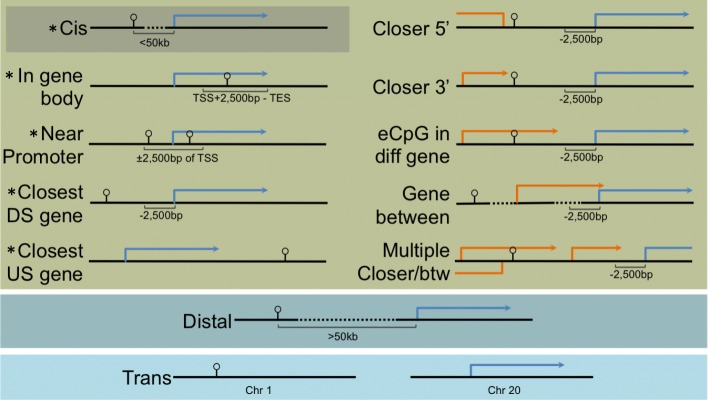
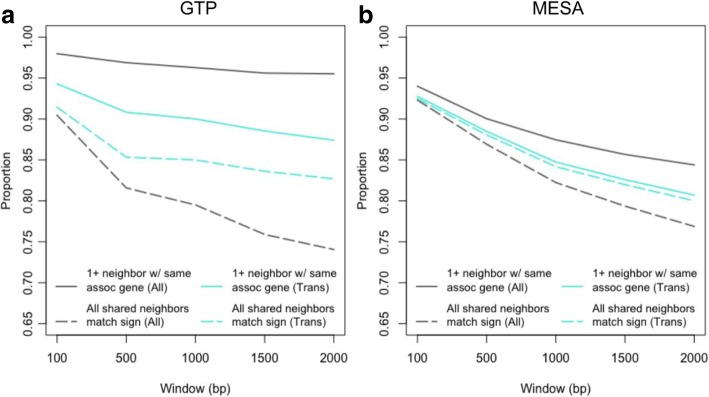
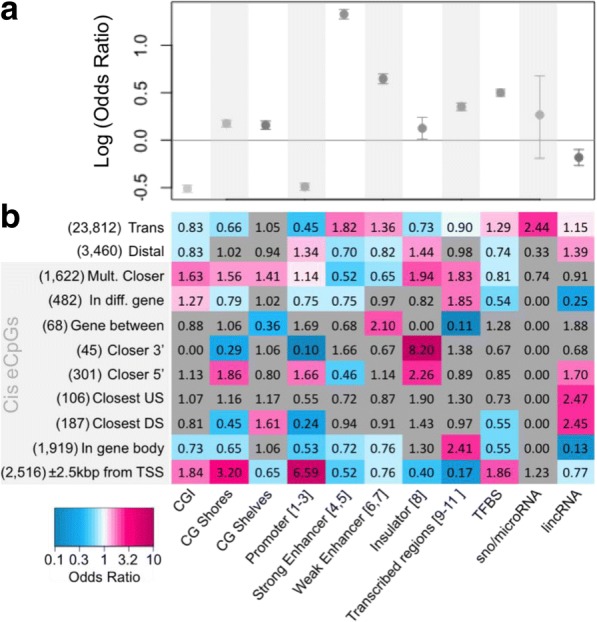
We used a linear mixed model framework to assess the correlation of DNA methylation at ~ 400 k CpGs with gene expression changes at ~ 13 k transcripts in two independent datasets from human blood cells. Among CpGs at which methylation significantly associates with transcription (eCpGs), > 50% are distal (> 50 kb) or trans (different chromosome) to the correlated gene. Many eCpG-transcript pairs are consistent between studies and ~ 90% of neighboring eCpGs associate with the same gene, within studies. We find that enhancers (P < 5e-18) and microRNA genes (P = 9e-3) are overrepresented among trans eCpGs, and insulators and long intergenic non-coding RNAs are enriched among cis and distal eCpGs. Intragenic-eCpG-transcript correlations are negative in 60-70% of occurrences and are enriched for annotated gene promoters and enhancers (P < 0.002), highlighting the importance of intragenic regulation. Gene Ontology analysis indicates that trans eCpGs are enriched for transcription factor genes and chromatin modifiers, suggesting that some trans eCpGs represent the influence of gene networks and higher-order transcriptional control.
This work sheds new light on the interplay between epigenetic changes and gene expression, and provides useful data for mining biologically-relevant results from epigenome-wide association studies.
基因表达可受到 DNA 甲基化的影响,这种影响既可以发生在远端的调控元件(如增强子)上,也可以发生在近端的启动子上。目前,我们对于远端 DNA 甲基化变化对基因表达模式的影响的了解并不完整。在这里,我们通过全基因组范围内的甲基化和表达模式来研究 DNA 甲基化与基因表达之间的相互作用,从而探索整个基因组中的情况。
我们使用线性混合模型框架,在来自人类血细胞的两个独立数据集的约 13000 个转录本中,评估了约 400000 个 CpG 位点的 DNA 甲基化与基因表达变化之间的相关性。在与转录显著相关的 CpG 中(eCpG),超过 50%的 CpG 位于相关基因的远端(>50kb)或跨(不同染色体)。在研究之间,许多 eCpG-转录本对是一致的,并且在研究内,约 90%的相邻 eCpG 与同一个基因相关。我们发现,增强子(P<5e-18)和 microRNA 基因(P=9e-3)在跨染色体的 eCpG 中过度表达,而绝缘子和长非编码 RNA 则在顺式和远端的 eCpG 中富集。在 60-70%的情况下,基因内 eCpG-转录本的相关性为负相关,并且在注释的基因启动子和增强子中富集(P<0.002),这突出了基因内调控的重要性。基因本体论分析表明,跨染色体的 eCpG 富集了转录因子基因和染色质修饰因子,这表明一些跨染色体的 eCpG 代表了基因网络和更高阶转录控制的影响。
这项工作为我们深入了解表观遗传变化与基因表达之间的相互作用提供了新的视角,并为从全基因组关联研究中挖掘具有生物学意义的结果提供了有用的数据。