Yu Jie, Zhao Jie, Song Yuqin, Zhang Jiachao, Yu Zhongjie, Zhang Heping, Sun Zhihong
Key Laboratory of Dairy Biotechnology and Engineering, Ministry of Education, Inner Mongolia Agricultural University, Hohhot, China.
Key Laboratory of Dairy Products Processing, Ministry of Agriculture, Inner Mongolia Agricultural University, Hohhot, China.
Front Microbiol. 2018 Jun 4;9:1151. doi: 10.3389/fmicb.2018.01151. eCollection 2018.
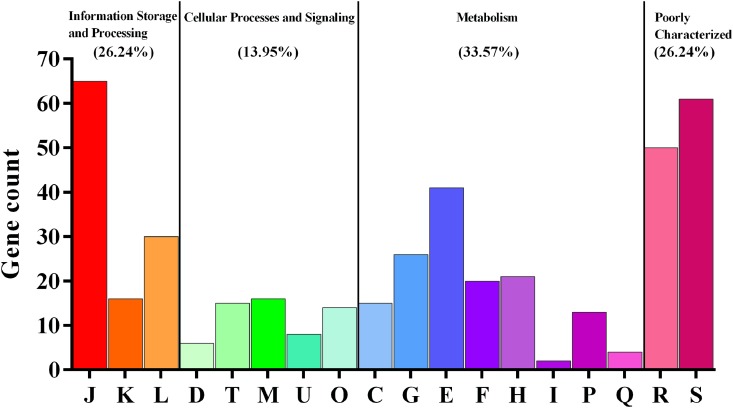
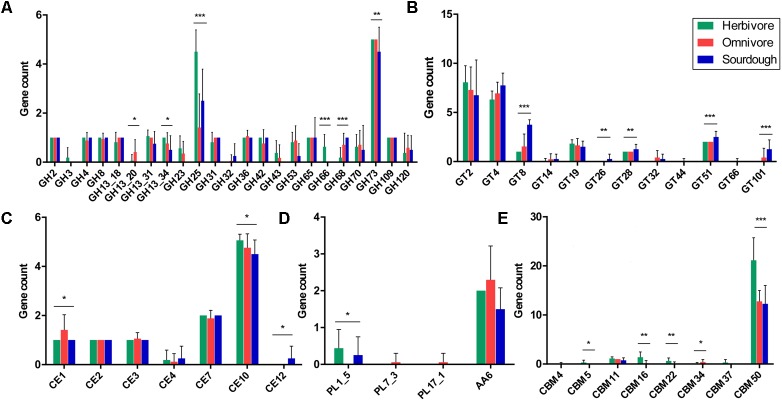
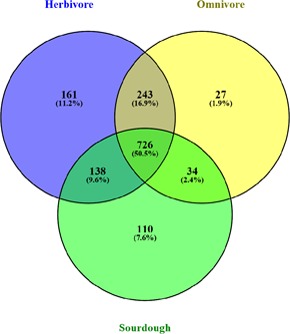
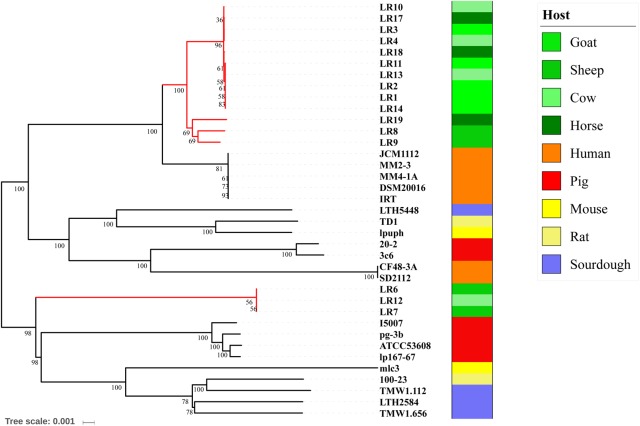
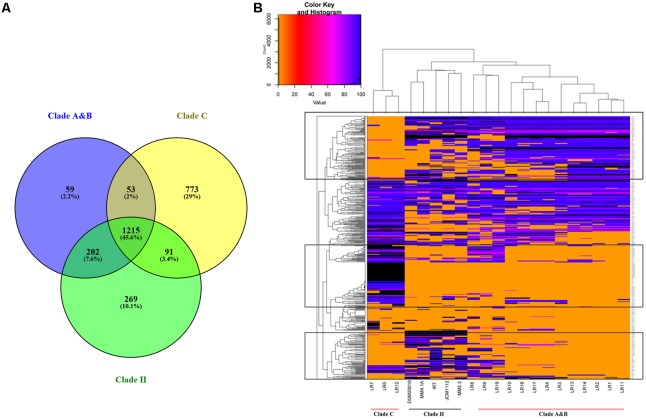
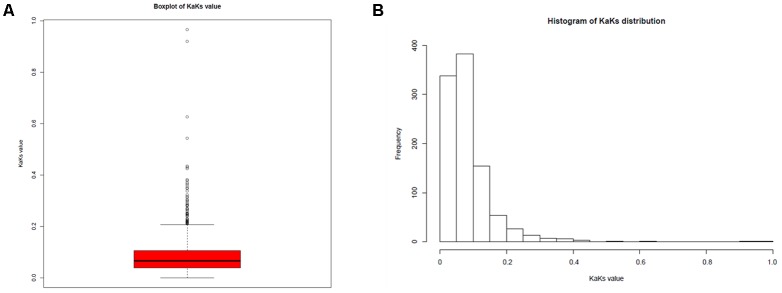
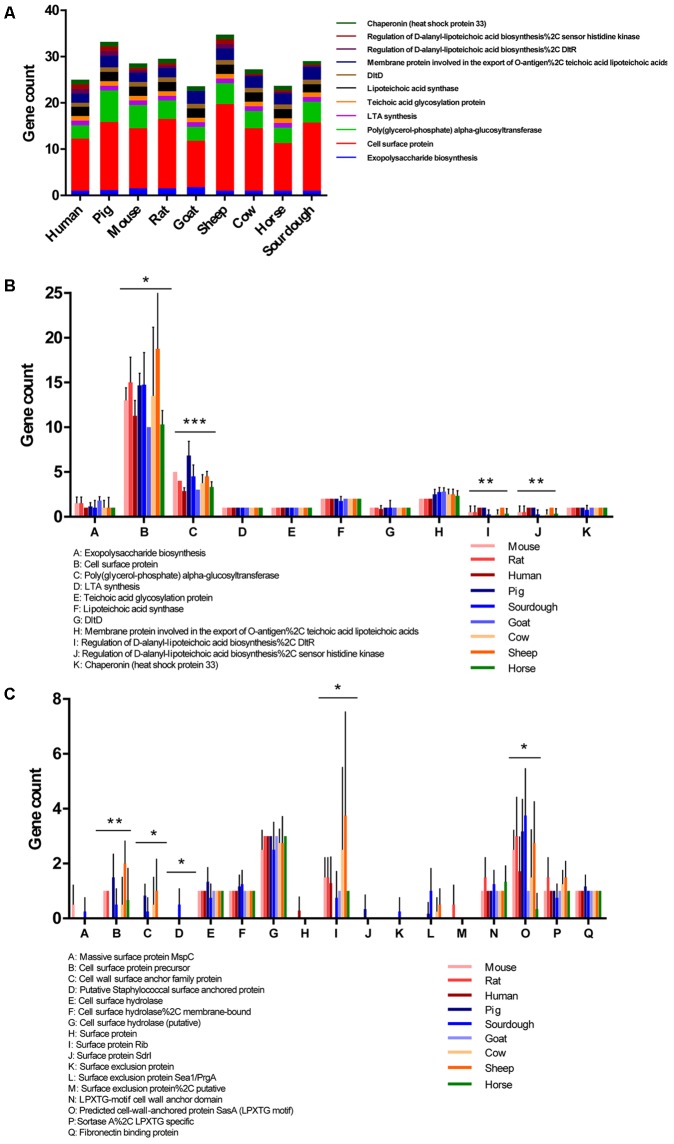
is a catalase-negative, Gram-positive, non-motile, obligately heterofermentative bacterial species that has been used as a model to describe the ecology and evolution of vertebrate gut symbionts. However, the genetic features and evolutionary strategies of from the gastrointestinal tract of herbivores remain unknown. Therefore, 16 strains isolated from goat, sheep, cow, and horse in Inner Mongolia, China were sequenced in this study. A comparative genomic approach was used to assess genetic diversity and gain insight into the distinguishing features related to the different hosts based on 21 published genomic sequences. Genome size, G + C content, and average nucleotide identity values of the strains from different hosts indicated that the strains have broad genetic diversity. The pan-genome of 37 strains contained 8,680 gene families, and the core genome contained 726 gene families. A total of 92,270 nucleotide mutation sites were discovered among 37 strains, and all core genes displayed a / ratio much lower than 1, suggesting strong purifying selective pressure (negative selection). A highly robust maximum likelihood tree based on the core genes shown in the herbivore isolates were divided into three clades; clades A and B contained most of the herbivore isolates and were more closely related to human isolates and vastly distinct from clade C. Some functional genes may be attributable to host-specific of the herbivore, omnivore, and sourdough groups. Moreover, the numbers of genes encoding cell surface proteins and active carbohydrate enzymes were host-specific. This study provides new insight into the adaptation of to the intestinal habitat of herbivores, suggesting that the genomic diversity of from different ecological origins is closely associated with their living environment.
是一种过氧化氢酶阴性、革兰氏阳性、不运动、专性异源发酵的细菌物种,已被用作描述脊椎动物肠道共生体的生态和进化的模型。然而,来自食草动物胃肠道的该物种的遗传特征和进化策略仍然未知。因此,本研究对从中国内蒙古的山羊、绵羊、牛和马中分离出的16株该物种进行了测序。基于21个已发表的基因组序列,采用比较基因组学方法评估遗传多样性,并深入了解与不同宿主相关的显著特征。来自不同宿主的该物种菌株的基因组大小、G + C含量和平均核苷酸同一性值表明这些菌株具有广泛的遗传多样性。37株该物种的泛基因组包含8680个基因家族,核心基因组包含726个基因家族。在37株该物种中总共发现了92270个核苷酸突变位点,并且所有核心基因显示出的非同义/同义替换率远低于1,表明存在强烈的纯化选择压力(负选择)。基于核心基因构建的高度稳健的最大似然树显示,食草动物分离株分为三个进化枝;进化枝A和B包含大多数食草动物分离株,与人类分离株关系更密切,与进化枝C截然不同。一些功能基因可能归因于食草动物、杂食动物和酸面团菌群的宿主特异性。此外,编码细胞表面蛋白和活性碳水化合物酶的基因数量具有宿主特异性。本研究为该物种适应食草动物肠道栖息地提供了新的见解,表明来自不同生态起源的该物种的基因组多样性与其生存环境密切相关。