Oslo Centre for Biostatistics and Epidemiology, Oslo University Hospital, Oslo universitetssykehus HF, Sogn Arena, PB 4950 Nydalen, Oslo, 0424, Norway.
MRC Biostatistics Unit, University of Cambridge, MRC Biostatistics Unit, Cambridge Institute of Public Health, Robinson Way, Cambridge, CB2 0SR, United Kingdom.
BMC Genomics. 2018 Jun 25;19(1):494. doi: 10.1186/s12864-018-4859-7.
There is considerable evidence that many complex traits have a partially shared genetic basis, termed pleiotropy. It is therefore useful to consider integrating genome-wide association study (GWAS) data across several traits, usually at the summary statistic level. A major practical challenge arises when these GWAS have overlapping subjects. This is particularly an issue when estimating pleiotropy using methods that condition the significance of one trait on the signficance of a second, such as the covariate-modulated false discovery rate (cmfdr).
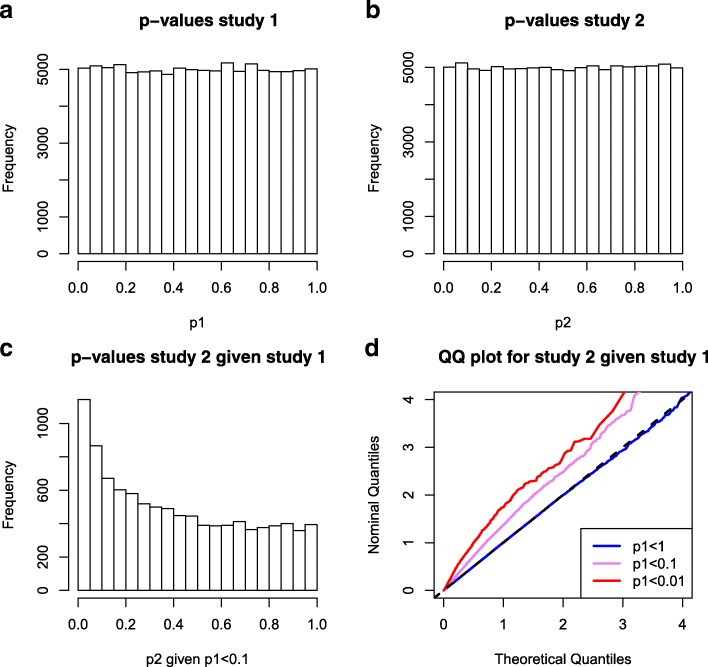
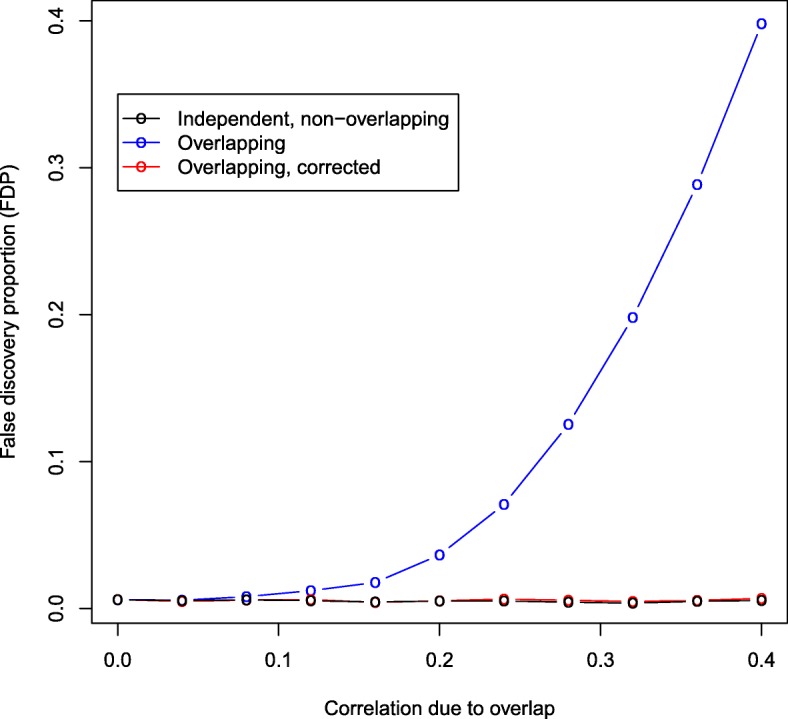
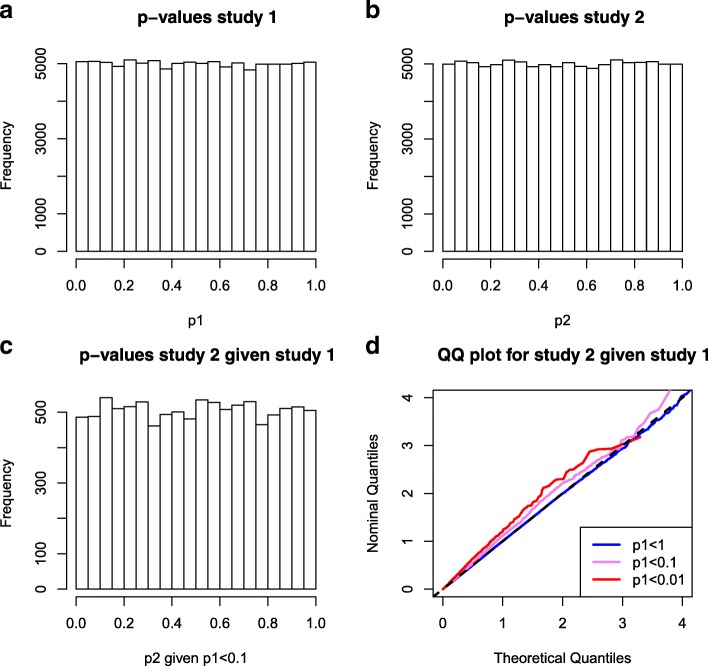
We propose a method for correcting for sample overlap at the summary statistic level. We quantify the expected amount of spurious correlation between the summary statistics from two GWAS due to sample overlap, and use this estimated correlation in a simple linear correction that adjusts the joint distribution of test statistics from the two GWAS. The correction is appropriate for GWAS with case-control or quantitative outcomes. Our simulations and data example show that without correcting for sample overlap, the cmfdr is not properly controlled, leading to an excessive number of false discoveries and an excessive false discovery proportion. Our correction for sample overlap is effective in that it restores proper control of the false discovery rate, at very little loss in power.
With our proposed correction, it is possible to integrate GWAS summary statistics with overlapping samples in a statistical framework that is dependent on the joint distribution of the two GWAS.
有相当多的证据表明,许多复杂的特征具有部分共同的遗传基础,称为多效性。因此,考虑整合多个特征的全基因组关联研究(GWAS)数据是很有用的,通常是在汇总统计数据水平上。当这些 GWAS 具有重叠的研究对象时,就会出现一个主要的实际挑战。当使用条件显著的方法估计多效性时,例如协变量调制的错误发现率(cmfdr),这尤其成问题。
我们提出了一种在汇总统计数据水平上校正样本重叠的方法。我们量化了由于样本重叠而导致两个 GWAS 汇总统计数据之间虚假相关性的预期数量,并在简单的线性校正中使用此估计相关性,该校正调整了两个 GWAS 的测试统计数据的联合分布。该校正适用于病例对照或定量结局的 GWAS。我们的模拟和数据示例表明,如果不校正样本重叠,cmfdr 就无法得到适当控制,导致大量的假发现和过高的假发现比例。我们的样本重叠校正有效地恢复了错误发现率的适当控制,而几乎没有损失功效。
通过我们提出的校正方法,可以在一个依赖于两个 GWAS 的联合分布的统计框架中整合具有重叠样本的 GWAS 汇总统计数据。