Ma Shuang, Schild Michael, Tran Diana, Zhang Xuefeng, Zhang Wan-Lin, Shen Shuai, Xu Hong-Tao, Yang Lian-He, Wang Endi
Department of Neurology, Sheng Jing Hospital of China Medical University, Shenyang, Liaoning, China Department of Pathology, Duke University Medical Center, Durham, NC Department of Pathology, First Affiliated Hospital and College of Basic Medical Sciences, China Medical University, Shenyang, Liaoning, China.
Medicine (Baltimore). 2018 Jun;97(26):e11271. doi: 10.1097/MD.0000000000011271.
Primary central nervous system histiocytic sarcoma (PCNSHS) is a rare lymphohematopoietic tumor with a histiocytic cell origin. To our knowledge, only 28 cases have been published in English and 2 cases in Chinese.
A 49-year-old Asian female presented to the hospital with a 2 month history of hypomnesia, odynophagia, and gait disorder. Physical examination demonstrated decreased lower extremity muscle strength. The patient denied a history of malignancy.
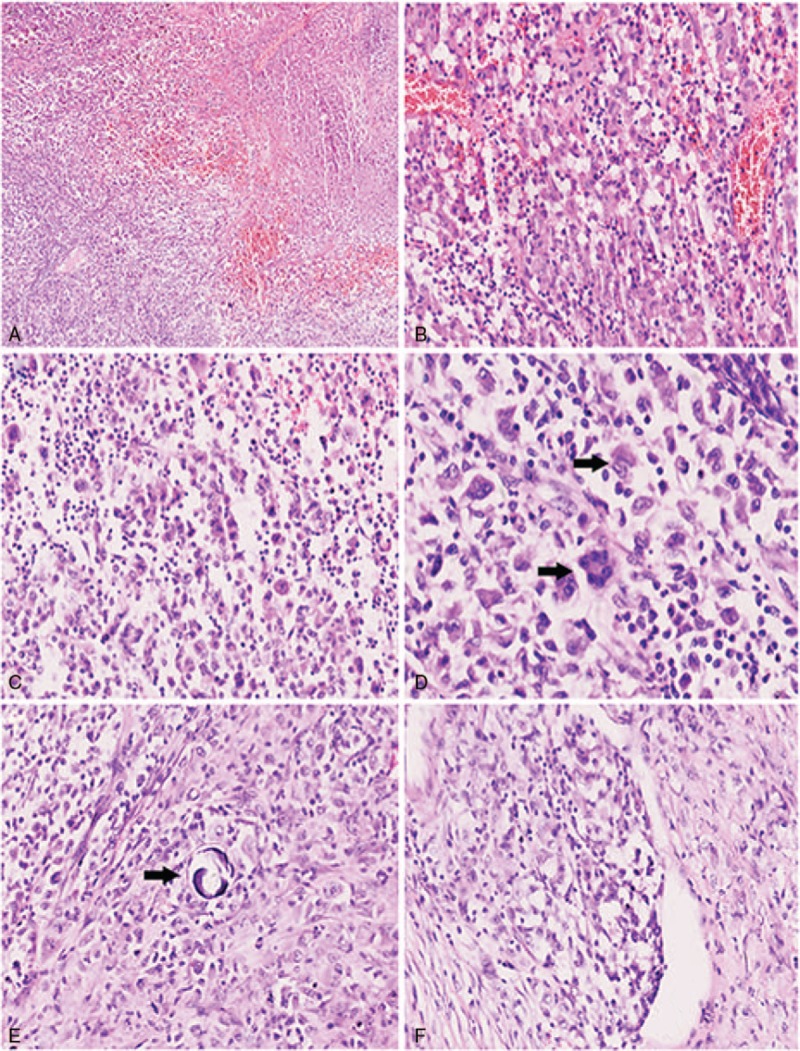
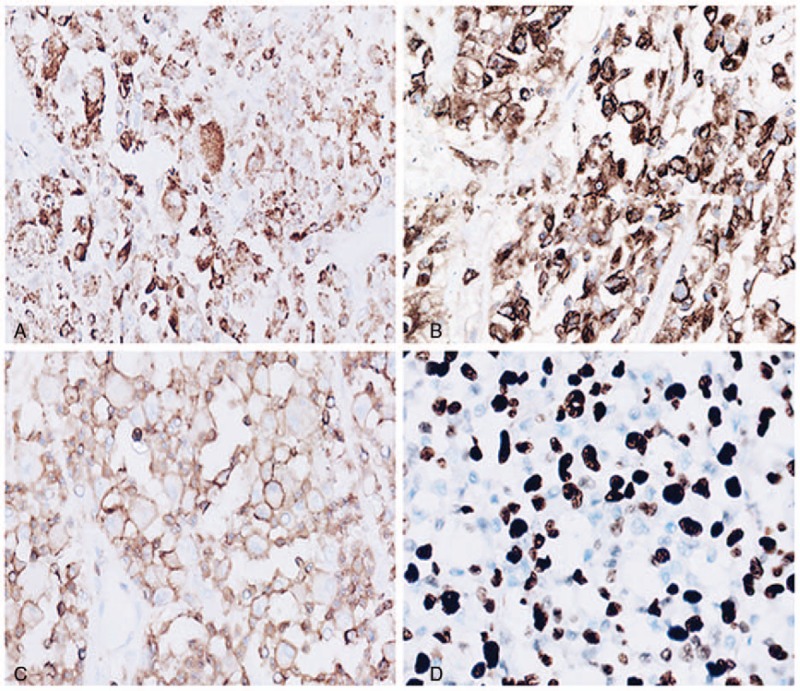
Radiology demonstrated a lesion in parietal lobe with uniformenhancement. Histologic analysis showed pleomorphic tumor cells with a loose arrangement, effacing the normal brain tissue. The tumor cells exhibited abundant eosinophilic cytoplasm, highly atypical nuclei and predominant nucleoli. Immunohistochemistry revealed positive immunoreactivity for CD45, lysozyme, CD68, and CD163, and negative for pan-cytokeratin (CK), epithelial membrane antigen (EMA), glial fibrillary acidic protein (GFAP), CD3, CD20, CD1a, CD79a, CD138, oligodendrocyte transcription factor (olig2), CD15, melan-A, CD30, CD21, CD35, Human Melanoma Black-45 (HMB45), and anaplastic lymphoma kinase-1 (ALK-1). The diagnosis of PCNSHS was rendered.
The patient underwent complete surgical resection and adjuvant radiotherapy.
Follow-up information shows the patient died 8 months following the initial diagnosis.
PCNSHS is extremely rare with an aggressive clinical course. Immunohistiochemistry is necessary to make this diagnosis and to exclude other primary intracranial and lymphohematopoietic tumors. Further research is required to improve the outcome of patients with PCNSHS.
原发性中枢神经系统组织细胞肉瘤(PCNSHS)是一种罕见的起源于组织细胞的淋巴造血肿瘤。据我们所知,英文文献仅发表了28例,中文文献发表了2例。
一名49岁的亚洲女性因记忆力减退、吞咽疼痛和步态障碍2个月就诊。体格检查显示下肢肌力下降。患者否认有恶性肿瘤病史。
影像学检查显示顶叶有一个均匀强化的病变。组织学分析显示多形性肿瘤细胞排列疏松,破坏了正常脑组织。肿瘤细胞表现为丰富的嗜酸性细胞质、高度异型性核和显著的核仁。免疫组化显示CD45、溶菌酶、CD68和CD163呈阳性免疫反应,而全细胞角蛋白(CK)、上皮膜抗原(EMA)、胶质纤维酸性蛋白(GFAP)、CD3、CD20、CD1a、CD79a、CD138、少突胶质细胞转录因子(olig2)、CD15、黑素A、CD30、CD21、CD35、人黑素瘤黑色45(HMB45)和间变性淋巴瘤激酶-1(ALK-1)呈阴性。诊断为PCNSHS。
患者接受了完整的手术切除和辅助放疗。
随访信息显示患者在初次诊断后8个月死亡。
PCNSHS极为罕见,临床病程侵袭性强。免疫组织化学对于做出此诊断和排除其他原发性颅内及淋巴造血肿瘤是必要的。需要进一步研究以改善PCNSHS患者的预后。