Postdoctoral Researcher, IBM Research, Cambridge, MA, 02142, USA.
Visiting Research Scientist, Mitsubishi Electric Research Laboratories, Cambridge, MA, 02139, USA.
Sci Rep. 2018 Jul 2;8(1):9936. doi: 10.1038/s41598-018-28364-3.
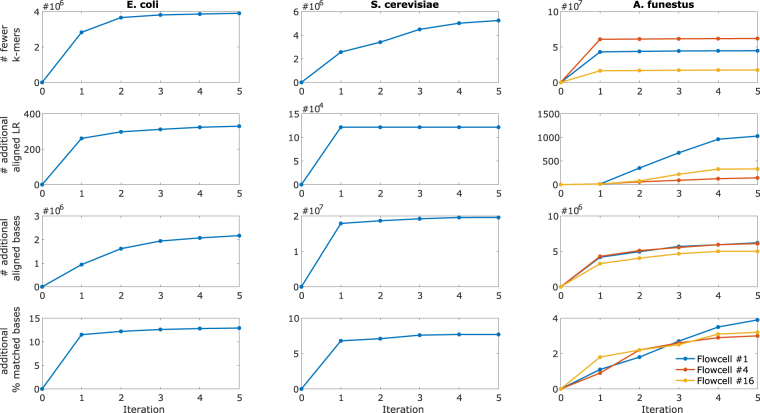
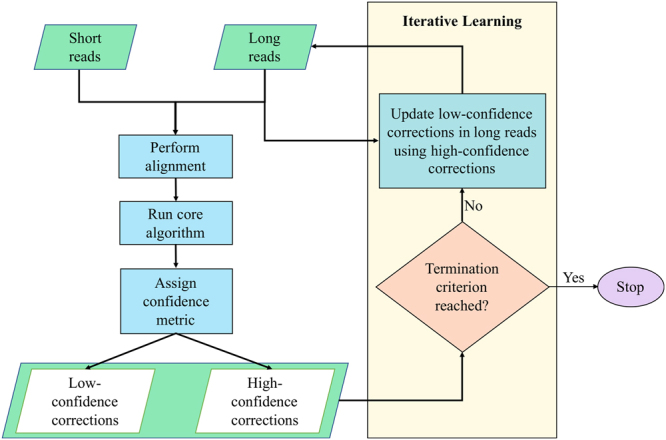
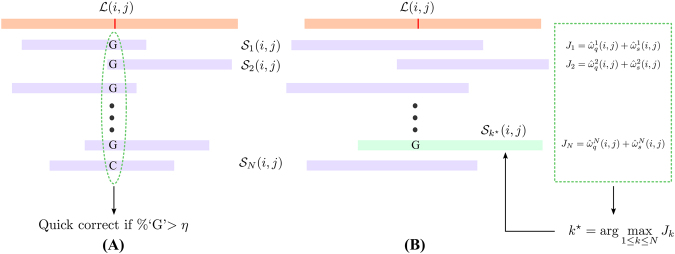
Second-generation DNA sequencing techniques generate short reads that can result in fragmented genome assemblies. Third-generation sequencing platforms mitigate this limitation by producing longer reads that span across complex and repetitive regions. However, the usefulness of such long reads is limited because of high sequencing error rates. To exploit the full potential of these longer reads, it is imperative to correct the underlying errors. We propose HECIL-Hybrid Error Correction with Iterative Learning-a hybrid error correction framework that determines a correction policy for erroneous long reads, based on optimal combinations of decision weights obtained from short read alignments. We demonstrate that HECIL outperforms state-of-the-art error correction algorithms for an overwhelming majority of evaluation metrics on diverse, real-world data sets including E. coli, S. cerevisiae, and the malaria vector mosquito A. funestus. Additionally, we provide an optional avenue of improving the performance of HECIL's core algorithm by introducing an iterative learning paradigm that enhances the correction policy at each iteration by incorporating knowledge gathered from previous iterations via data-driven confidence metrics assigned to prior corrections.
第二代 DNA 测序技术产生的短读长会导致基因组组装片段化。第三代测序平台通过生成跨越复杂和重复区域的长读长来缓解这一限制。然而,由于测序错误率高,这种长读长的用处有限。为了充分利用这些更长的读长,必须纠正潜在的错误。我们提出了 HECIL-Hybrid Error Correction with Iterative Learning,这是一种混合错误校正框架,它基于从短读对齐中获得的最优决策权重组合,为错误的长读确定校正策略。我们证明,在包括大肠杆菌、酿酒酵母和疟疾病媒按蚊 A. funestus 在内的各种真实世界数据集上,HECIL 在绝大多数评估指标上都优于最先进的错误校正算法。此外,我们通过引入迭代学习范例提供了一种改进 HECIL 核心算法性能的可选途径,该范例通过使用数据驱动的置信度指标将从前几次迭代中收集到的知识分配给先前的校正,从而在每次迭代中增强校正策略。