Jouhadi Zineb, Odou Marie Francoise, Zerimech Farid, Bousfiha Ahmed Aziz, Mikou Nabiha, Porchet Nicole, Crepin Michel, Najib Jilali, Balduyck Malika
Pediatric Infectious Diseases Department, Faculty of Medicine and Pharmacy Hassan II University, Casablanca, Morocco.
CHU Lille, Service de Biochimie et Biologie Moléculaire Hormonologie, Métabolisme-Nutrition, Oncologie, F-59000 Lille, France.
Respir Med Case Rep. 2018 Apr 10;24:58-62. doi: 10.1016/j.rmcr.2018.04.005. eCollection 2018.
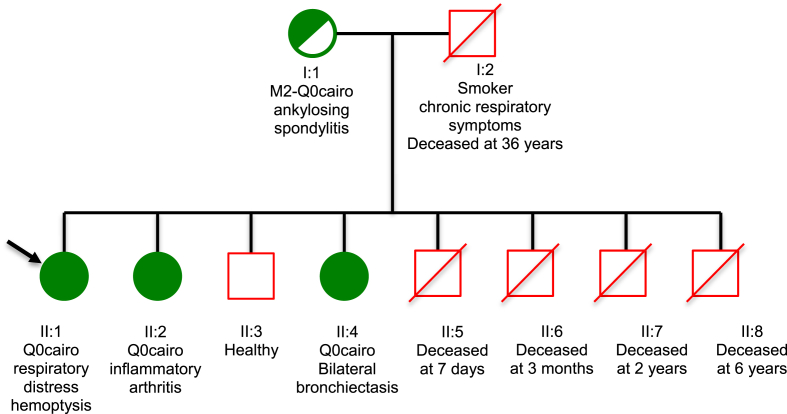
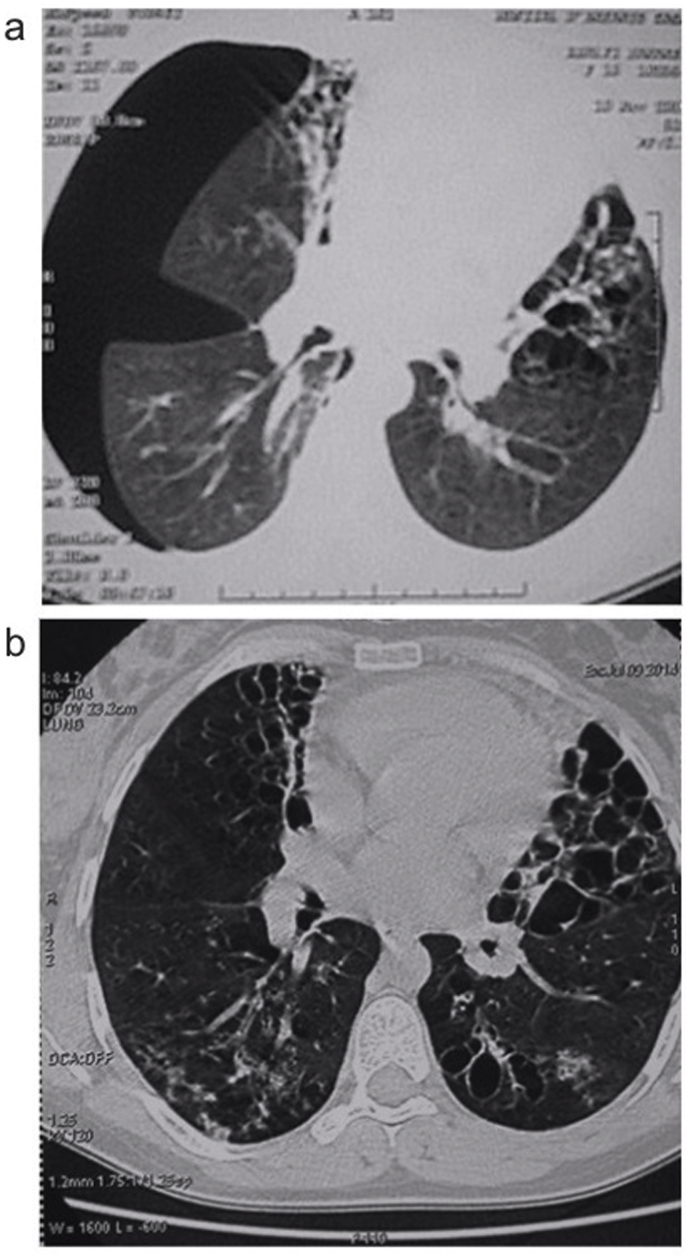
Alpha-1 antitrypsin deficiency is an autosomal, codominant disorder caused by mutations of the gene. This genetic disorder is mainly associated with development of pulmonary emphysema and/or chronic liver disease and cirrhosis. Here we report a very rare alpha-1 antitrypsin Null Q0cairo homozygous mutation characterized by a complete absence of alpha-1 antitrypsin in the plasma, in a non-consanguineous Moroccan family. This mutation has been previously described in heterozygosis in only three cases worldwide: an Italian/Egyptian family and two Italian families (Zorzetto et al., 2005). The main clinical features in two members of this Moroccan family were the severity and precocity of bronchiectasis, quickly spreading and seriously limiting respiratory function and physical activity by the second decade of age. Moreover, the index case presented with many episodes of pulmonary infections concomitant with severe neutropenia. The third member of the family presented with ankylosing spondyloarthritis and developed panniculitis later but had no respiratory symptoms. The presence of this alpha-1-antitrypsin Q0cairo homozygous mutation could explain the severity of clinical manifestations. Moreover, our observations highlight a great variability of clinical expression for the same mutation: early severe bronchiectasis, panniculitis, rheumatologic manifestations. This study further underlines the importance of genotyping by whole gene sequencing in addition to serum alpha-1 antitrypsin determination, to enable detection of alpha-1 antitrypsin deficiency due to rare genotypes.
α-1抗胰蛋白酶缺乏症是一种由该基因突变引起的常染色体共显性疾病。这种遗传疾病主要与肺气肿和/或慢性肝病及肝硬化的发展相关。在此,我们报告了一个非近亲结婚的摩洛哥家庭中一种非常罕见的α-1抗胰蛋白酶Null Q0开罗纯合突变,其特征是血浆中完全缺乏α-1抗胰蛋白酶。这种突变此前仅在全球三例杂合子病例中被描述过:一个意大利/埃及家庭和两个意大利家庭(佐尔泽托等人,2005年)。这个摩洛哥家庭的两名成员的主要临床特征是支气管扩张的严重程度和早熟性,在第二个十年时迅速蔓延并严重限制呼吸功能和身体活动。此外,索引病例出现多次肺部感染并伴有严重中性粒细胞减少症。该家庭的第三名成员患有强直性脊柱炎,后来发展为脂膜炎,但没有呼吸道症状。这种α-1抗胰蛋白酶Q0开罗纯合突变的存在可以解释临床表现的严重程度。此外,我们的观察结果突出了同一突变临床表达的巨大变异性:早期严重支气管扩张、脂膜炎、风湿病表现。这项研究进一步强调了除血清α-1抗胰蛋白酶测定外,通过全基因测序进行基因分型的重要性,以便能够检测由于罕见基因型导致的α-1抗胰蛋白酶缺乏症。