Sztupinszki Zsofia, Diossy Miklos, Krzystanek Marcin, Reiniger Lilla, Csabai István, Favero Francesco, Birkbak Nicolai J, Eklund Aron C, Syed Ali, Szallasi Zoltan
1Department of Bio and Health Informatics, Technical University of Denmark, Kemitorvet 208, Lyngby, 2800 Denmark.
21st Department of Pathology and Experimental Research, Semmelweis University, Budapest, Hungary.
NPJ Breast Cancer. 2018 Jul 2;4:16. doi: 10.1038/s41523-018-0066-6. eCollection 2018.
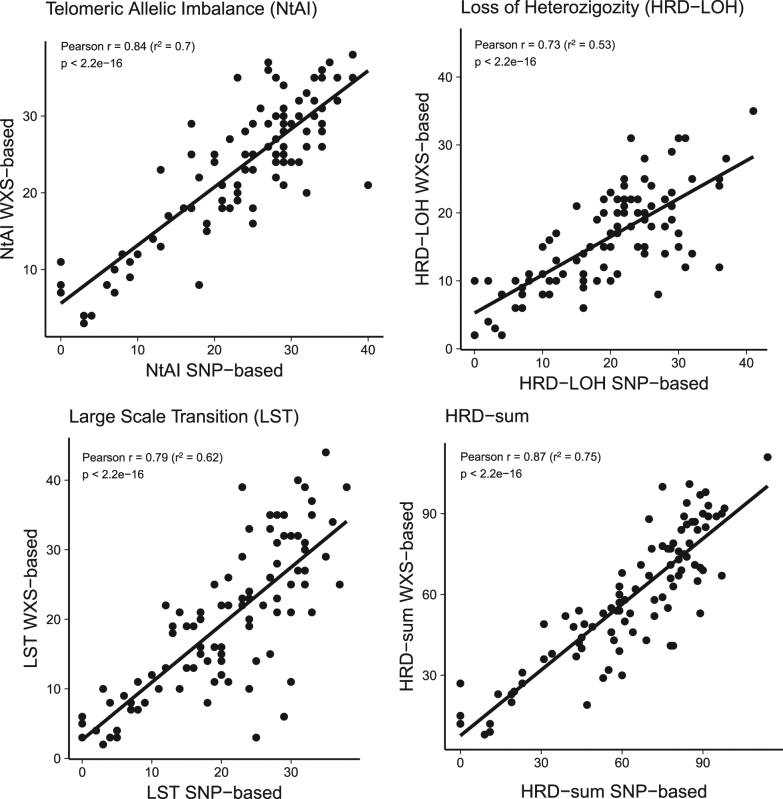
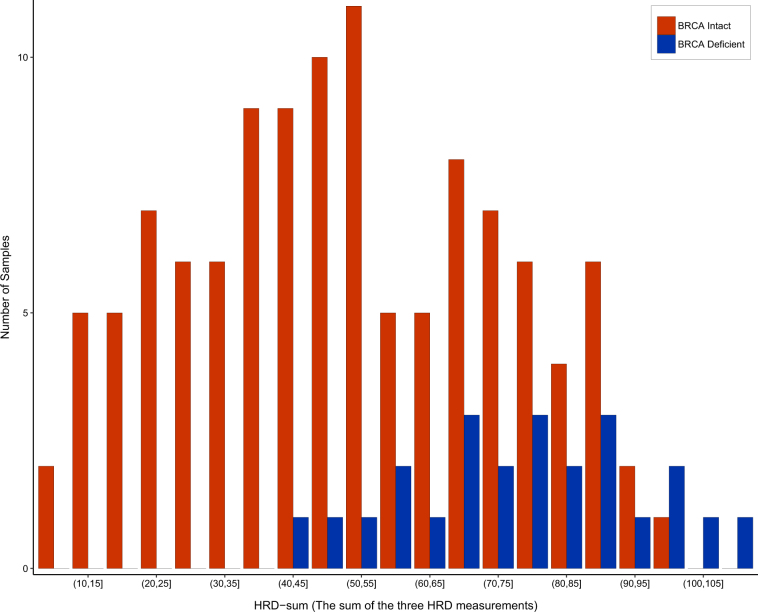
The first genomic scar-based homologous recombination deficiency (HRD) measures were produced using SNP arrays. As array-based technology has been largely replaced by next generation sequencing approaches, it has become important to develop algorithms that derive the same type of genomic scar scores from next generation sequencing (whole exome "WXS", whole genome "WGS") data. In order to perform this analysis, we introduce here the scarHRD R package and show that using this method the SNP array-based and next generation sequencing-based derivation of HRD scores show good correlation (Pearson correlation between 0.73 and 0.87 depending on the actual HRD measure) and that the NGS-based HRD scores distinguish similarly well between BRCA mutant and BRCA wild-type cases in a cohort of triple-negative breast cancer patients of the TCGA data set.
最初基于基因组疤痕的同源重组缺陷(HRD)测量是使用单核苷酸多态性(SNP)阵列进行的。由于基于阵列的技术已在很大程度上被新一代测序方法所取代,因此开发能够从新一代测序(全外显子组“WXS”、全基因组“WGS”)数据中得出相同类型基因组疤痕评分的算法变得至关重要。为了进行此分析,我们在此引入scarHRD R软件包,并表明使用此方法,基于SNP阵列和基于新一代测序得出的HRD评分显示出良好的相关性(根据实际的HRD测量,皮尔逊相关性在0.73至0.87之间),并且在TCGA数据集的三阴性乳腺癌患者队列中,基于新一代测序的HRD评分在区分BRCA突变型和BRCA野生型病例方面表现同样出色。