Alkek Center for Metagenomics and Microbiome Research, Baylor College of Medicine, Houston, TX, 77030, USA.
Department of Molecular Virology and Microbiology, Baylor College of Medicine, Houston, TX, 77030, USA.
Nat Commun. 2018 Aug 10;9(1):3205. doi: 10.1038/s41467-018-05658-8.
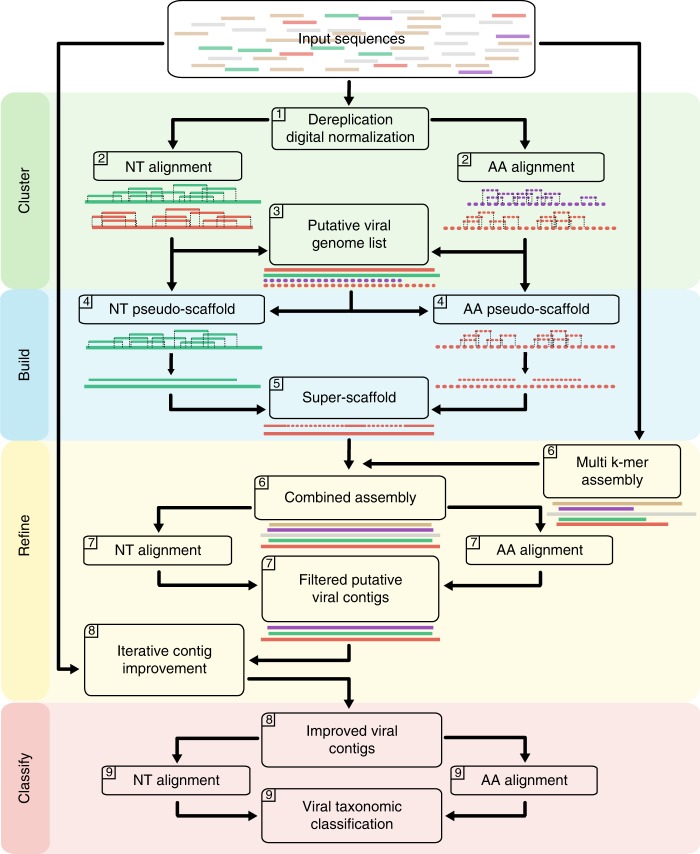
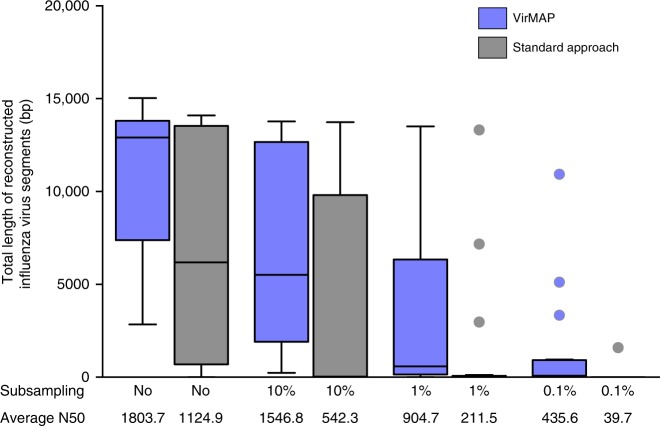
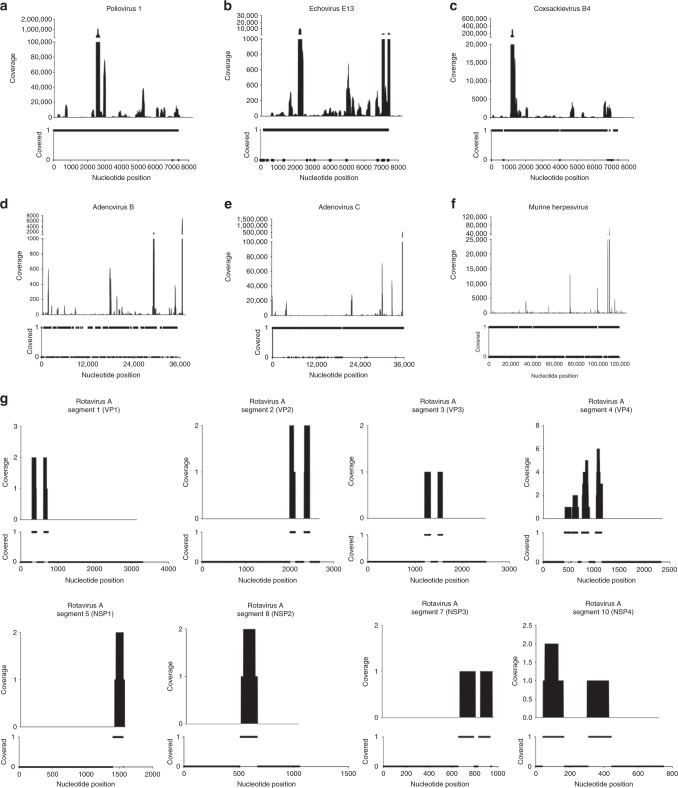
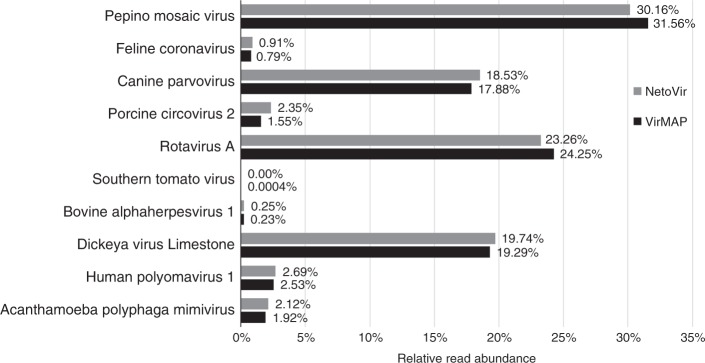
Accurate classification of the human virome is critical to a full understanding of the role viruses play in health and disease. This implies the need for sensitive, specific, and practical pipelines that return precise outputs while still enabling case-specific post hoc analysis. Viral taxonomic characterization from metagenomic data suffers from high background noise and signal crosstalk that confounds current methods. Here we develop VirMAP that overcomes these limitations using techniques that merge nucleotide and protein information to taxonomically classify viral reconstructions independent of genome coverage or read overlap. We validate VirMAP using published data sets and viral mock communities containing RNA and DNA viruses and bacteriophages. VirMAP offers opportunities to enhance metagenomic studies seeking to define virome-host interactions, improve biosurveillance capabilities, and strengthen molecular epidemiology reporting.
准确分类人类病毒组对于充分了解病毒在健康和疾病中的作用至关重要。这意味着需要敏感、特异和实用的管道,既能返回精确的输出,又能支持特定于病例的事后分析。从宏基因组数据中进行病毒分类特征分析受到高背景噪声和信号串扰的影响,这使得当前的方法变得复杂。在这里,我们开发了 VirMAP,它使用融合核苷酸和蛋白质信息的技术来克服这些限制,从而在不考虑基因组覆盖度或读取重叠的情况下对病毒重建体进行分类学分类。我们使用已发表的数据集和包含 RNA 和 DNA 病毒以及噬菌体的病毒模拟群落来验证 VirMAP。VirMAP 为寻求定义病毒组-宿主相互作用、提高生物监测能力和加强分子流行病学报告的宏基因组研究提供了机会。