Dong Zhiyu, Wang Junwen, Zhan Tingting, Xu Shuchang
Department of Gastroenterology, Tongji Hospital, Tongji University School of Medicine, Shanghai, China,
Onco Targets Ther. 2018 Jul 25;11:4327-4337. doi: 10.2147/OTT.S156716. eCollection 2018.
Esophageal adenocarcinoma (EAC) is the most common type of esophageal cancer in Western countries. It is usually detected at an advanced stage and has a poor prognosis. The aim of this study was to identify key genes and miRNAs in EAC.
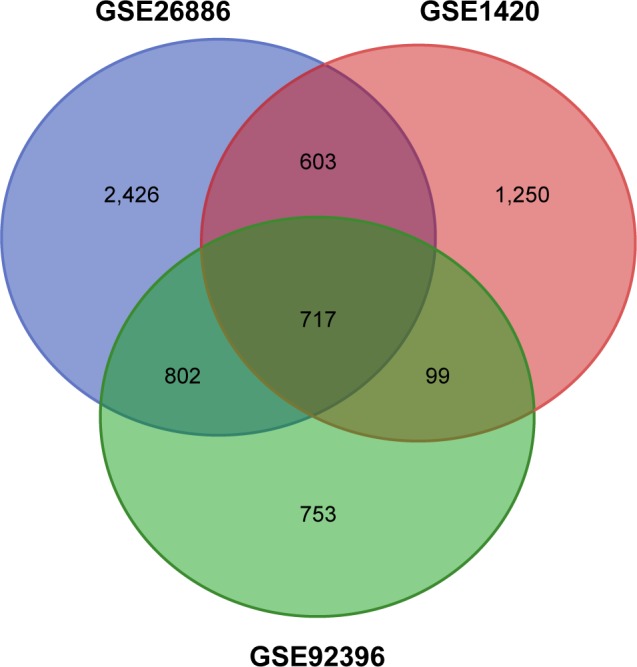
The mRNA microarray data sets GSE1420, GSE26886, and GSE92396 and miRNA data set GSE16456 were downloaded from the Gene Expression Omnibus database. Differentially expressed genes (DEGs) and differentially expressed miRNAs (DEMs) were obtained using R software. Functional enrichment analysis was performed using the DAVID database. A protein-protein interaction (PPI) network and functional modules were established using the STRING database and visualized by Cytoscape. The targets of the DEMs were predicted using the miRecords database, and overlapping genes between DEGs and targets were identified. The prognosis-related overlapping genes were identified using Kaplan-Meier analysis and Cox proportional hazard analysis based on The Cancer Genome Atlas (TCGA) database. The differential expression of these prognosis-related genes was validated using the expression matrix in the TCGA database.
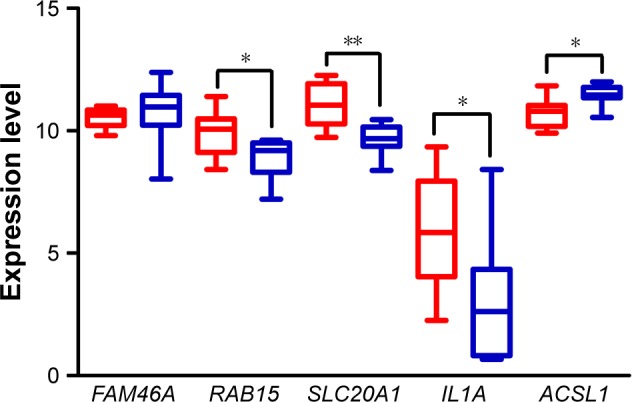
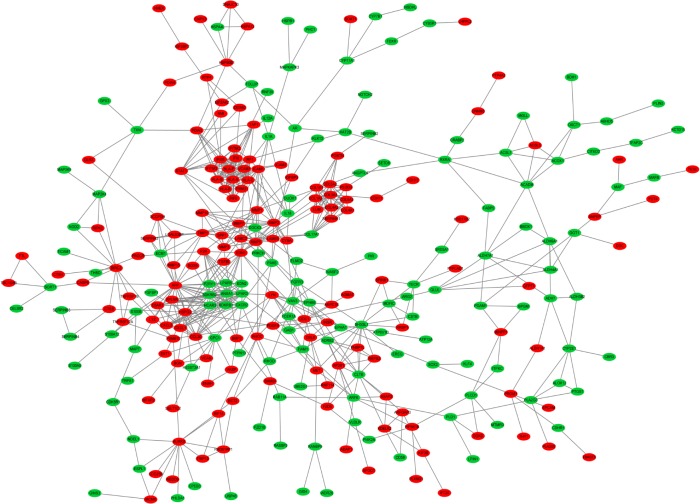
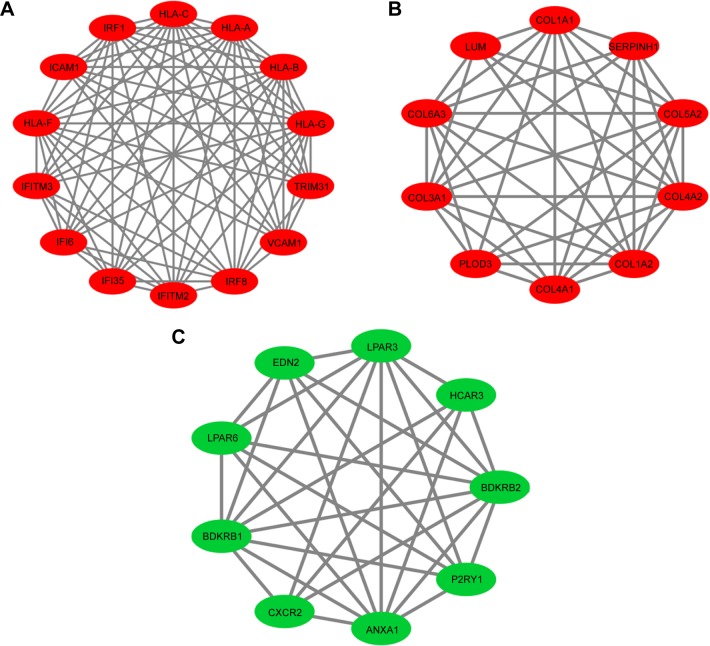
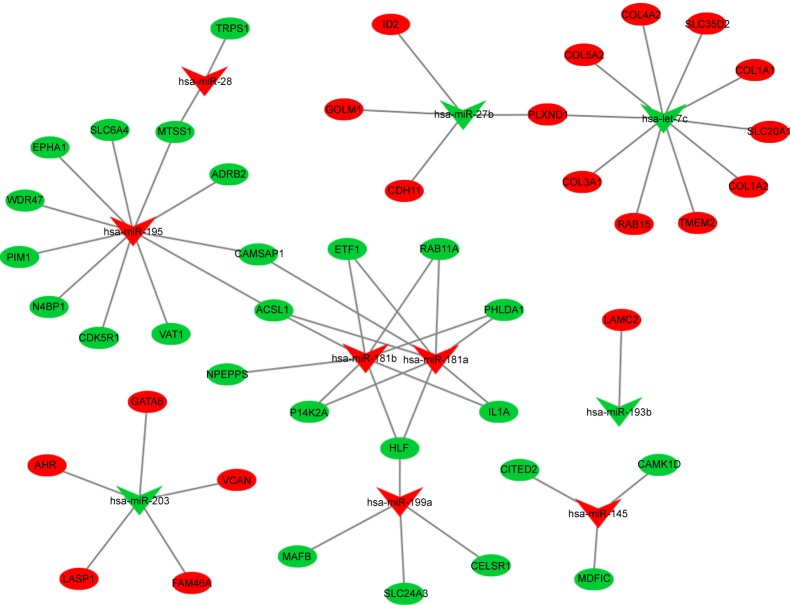
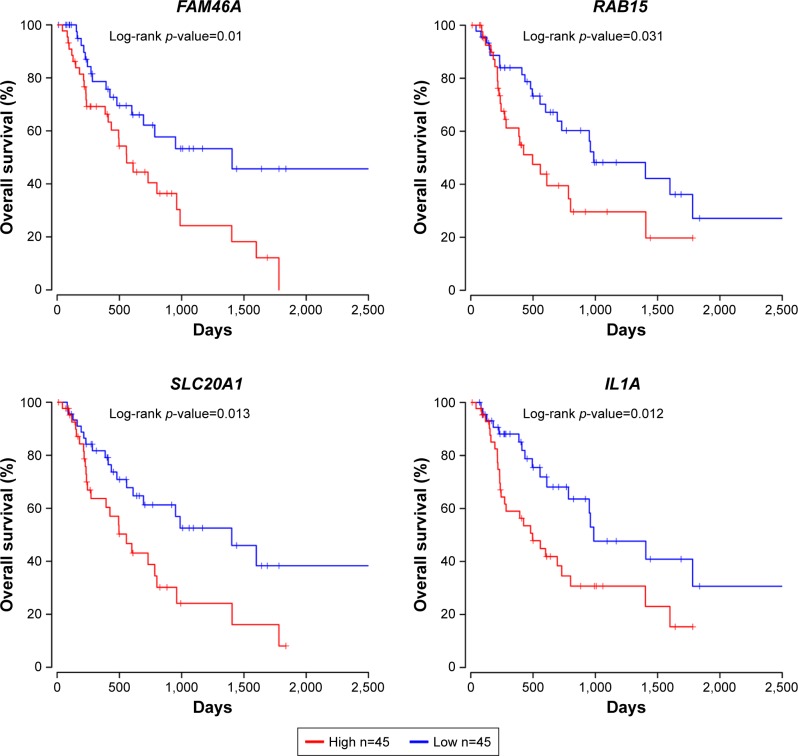
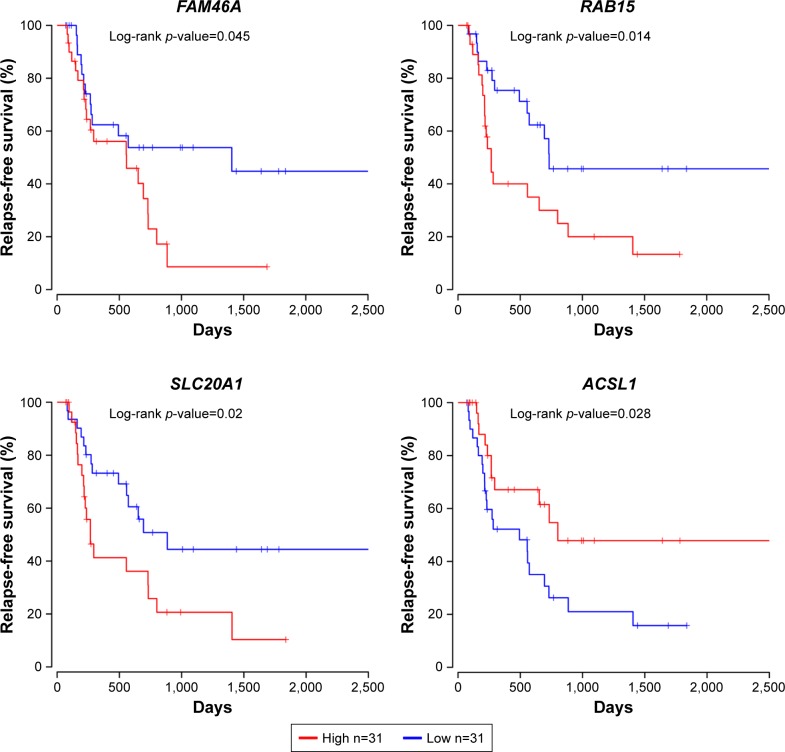
Seven hundred and fifteen DEGs were obtained, consisting of 313 upregulated and 402 downregulated genes. The PPI network consisted of 281 nodes; 683 edges were constructed and 3 functional modules were established. Forty-four overlapping genes and 56 miRNA- mRNA pairs were identified. Five genes, , , , , and , were associated with overall survival or relapse-free survival. and were found to be independent prognostic indicators for overall survival, and , , and were considered independent prognostic indicators for relapse-free survival. Among them, the overexpression of and and lower expression of were also identified in EAC tissues based on the expression matrix in the TCGA database.
These prognosis-related genes and differentially expressed miRNA have provided potential biomarkers for EAC diagnosis and treatment.
食管腺癌(EAC)是西方国家最常见的食管癌类型。它通常在晚期被检测到,预后较差。本研究的目的是鉴定EAC中的关键基因和微小RNA(miRNA)。
从基因表达综合数据库下载mRNA微阵列数据集GSE1420、GSE26886和GSE92396以及miRNA数据集GSE16456。使用R软件获得差异表达基因(DEG)和差异表达miRNA(DEM)。使用DAVID数据库进行功能富集分析。使用STRING数据库建立蛋白质-蛋白质相互作用(PPI)网络和功能模块,并通过Cytoscape进行可视化。使用miRecords数据库预测DEM的靶标,并鉴定DEG与靶标之间的重叠基因。基于癌症基因组图谱(TCGA)数据库,使用Kaplan-Meier分析和Cox比例风险分析鉴定与预后相关的重叠基因。使用TCGA数据库中的表达矩阵验证这些与预后相关基因的差异表达。
共获得715个DEG,其中313个基因上调,402个基因下调。PPI网络由281个节点组成;构建了683条边,并建立了3个功能模块。鉴定出44个重叠基因和56个miRNA- mRNA对。5个基因, 、 、 、 和 ,与总生存期或无复发生存期相关。 和 被发现是总生存期的独立预后指标, 、 和 被认为是无复发生存期的独立预后指标。其中,基于TCGA数据库中的表达矩阵,在EAC组织中也鉴定出 和 的过表达以及 的低表达。
这些与预后相关的基因和差异表达的miRNA为EAC的诊断和治疗提供了潜在的生物标志物。