von Manstein V, Groner B
Georg Speyer Haus , Institute for Tumor Biology and Experimental Therapy , Paul Ehrlich Str. 42 , 60596 Frankfurt am Main , Germany . Email:
Medchemcomm. 2016 Oct 14;8(1):96-102. doi: 10.1039/c6md00463f. eCollection 2017 Jan 1.
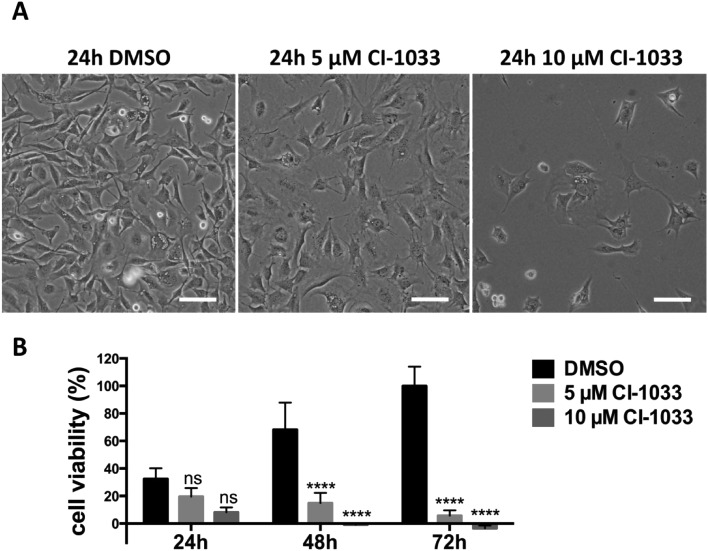
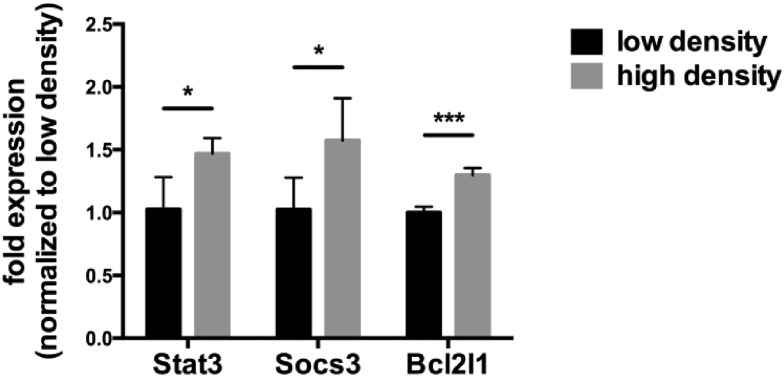
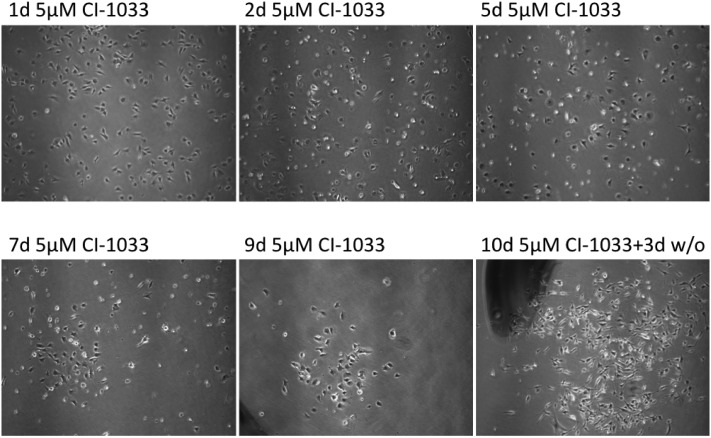
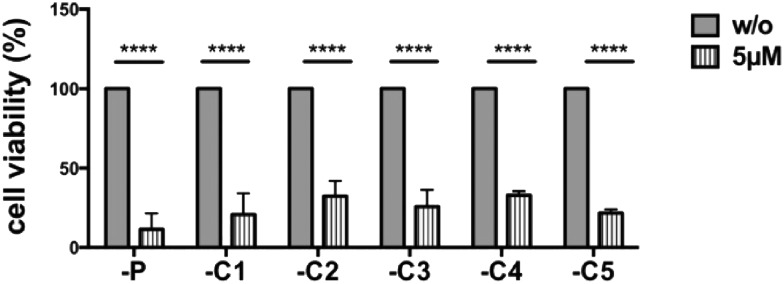
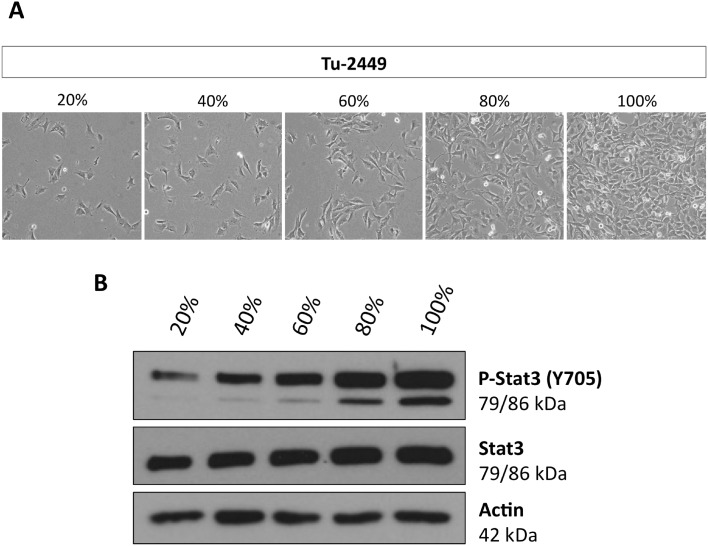
Tumor cell resistance to drug treatment severely limits the therapeutic success of treatment. Tumor cells, exposed to chemotherapeutic drugs, have developed intricate strategies to escape the cytotoxic effects and adapt to adverse conditions. The molecular mechanisms causing drug resistance can be based upon modifications of drug transport or metabolism, structural alterations of drug targets or adaptation of cellular signaling. An important component in the transformation of cells and the emergence of drug resistance is the activation of the transcription factor Stat3. The persistent, inappropriate activation of Stat3 causes the expression of target genes which promote tumor cell proliferation, survival, invasion and immune suppression, and it is instrumental in the process of the emergence of resistance to both conventional chemotherapeutic agents and novel targeted compounds. For these reasons, Stat3 inhibition is being pursued as a promising therapeutic strategy. We have investigated the effects of the tyrosine kinase inhibitor canertinib on the glioma cell line Tu-2449. In these cells Stat3 is persistently phosphorylated and activated downstream of the oncogenic driver v-Src and its effector, the cytoplasmic tyrosine kinase Bmx. Canertinib exposure of Tu-2449 cells rapidly caused the inhibition of the Bmx kinase and the deactivation of Stat3. Prolonged exposure of the cells to canertinib caused the death of the large majority of the cells. Only a few cells became resistant to canertinib and survived in tight clusters. These cells have become drug resistant. When the canertinib resistant cells were expanded and cultured at lower cell densities, they regained their sensitivity towards canertinib. We measured the extent of Stat3 activation as a function of cell density and found that higher cell densities are accompanied by increased Stat3 activation and a higher expression of Stat3 target genes. We suggest that Stat3 induction through tight cell-cell interactions, most likely through the engagement of cadherins, can counteract the inhibitory effects exerted by canertinib on Bmx. Cell-cell interactions induced Stat3 and compensated for the suppression of Stat3 by canertinib, thus transiently protecting the cells from the cytotoxic effects of the inhibitor.
肿瘤细胞对药物治疗的耐药性严重限制了治疗的成功。暴露于化疗药物的肿瘤细胞已形成复杂的策略来逃避细胞毒性作用并适应不利条件。导致耐药性的分子机制可能基于药物转运或代谢的改变、药物靶点的结构改变或细胞信号传导的适应。细胞转化和耐药性出现的一个重要组成部分是转录因子Stat3的激活。Stat3的持续、不适当激活导致促进肿瘤细胞增殖、存活、侵袭和免疫抑制的靶基因表达,并且在对传统化疗药物和新型靶向化合物产生耐药性的过程中起作用。由于这些原因,抑制Stat3被视为一种有前景的治疗策略。我们研究了酪氨酸激酶抑制剂卡奈替尼对胶质瘤细胞系Tu-2449的影响。在这些细胞中,Stat3在致癌驱动因子v-Src及其效应物细胞质酪氨酸激酶Bmx的下游持续磷酸化并被激活。用卡奈替尼处理Tu-2449细胞会迅速导致Bmx激酶的抑制和Stat3的失活。细胞长时间暴露于卡奈替尼会导致绝大多数细胞死亡。只有少数细胞对卡奈替尼产生耐药性并紧密聚集存活下来。这些细胞已产生耐药性。当将卡奈替尼耐药细胞以较低细胞密度扩增培养时,它们恢复了对卡奈替尼的敏感性。我们测量了Stat3激活程度与细胞密度的关系,发现较高的细胞密度伴随着Stat3激活增加和Stat3靶基因表达升高。我们认为,通过紧密的细胞间相互作用诱导Stat3,最有可能是通过钙黏蛋白的结合,可以抵消卡奈替尼对Bmx的抑制作用。细胞间相互作用诱导Stat3并补偿卡奈替尼对Stat3的抑制,从而暂时保护细胞免受抑制剂的细胞毒性作用。