Division of Nuclear Medicine and Molecular Imaging, Massachusetts General Hospital & Department of Radiology, Harvard Medical School, Boston, MA, 02114, USA; Department of Chemistry, School of Science, Tianjin University, 92 Weijin Road, Nankai District, Tianjin 300072, China.
Department of Radiopharmaceutics Development, National Institute of Radiological Sciences, National Institutes for Quantum and Radiological Science and Technology, Chiba, 263-8555, Japan.
Eur J Med Chem. 2018 Sep 5;157:898-908. doi: 10.1016/j.ejmech.2018.08.019. Epub 2018 Aug 9.
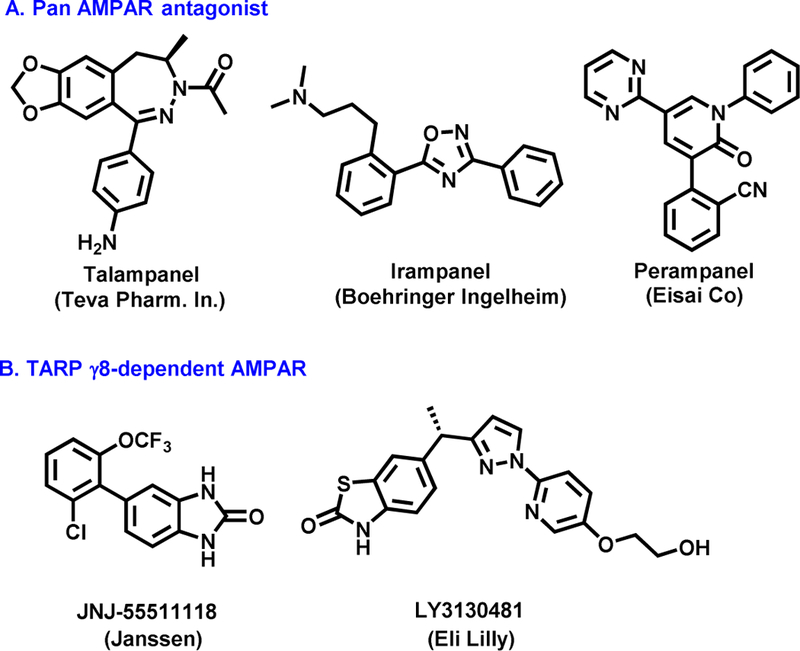
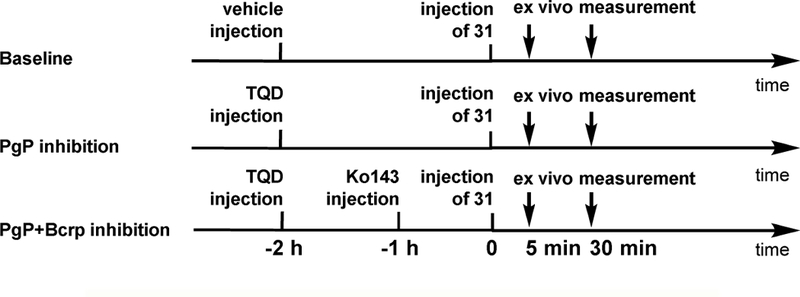
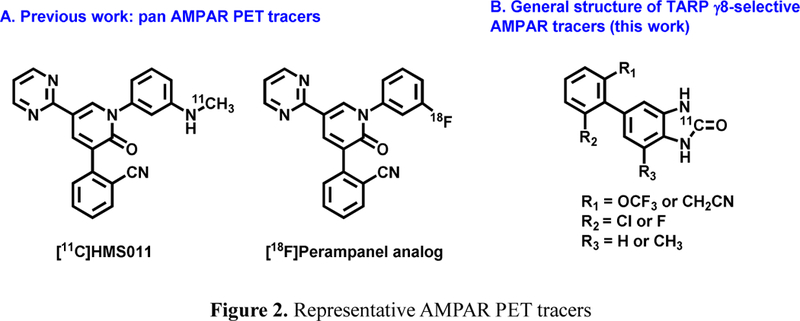
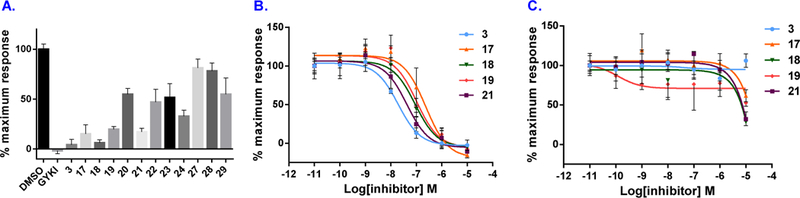
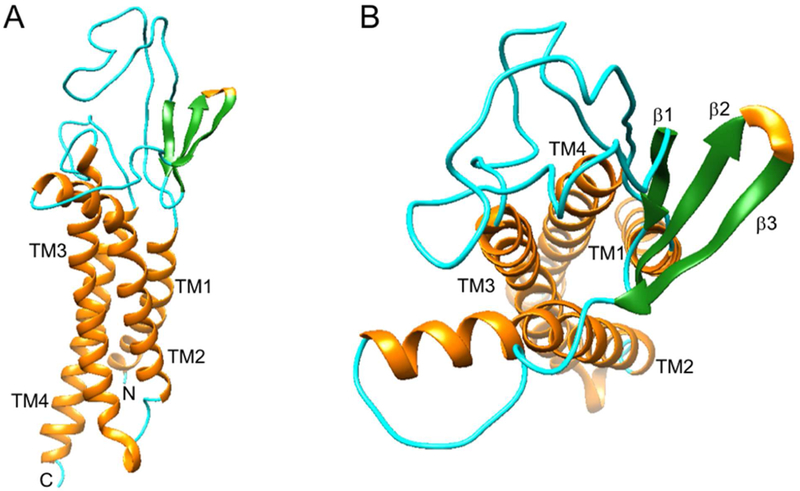
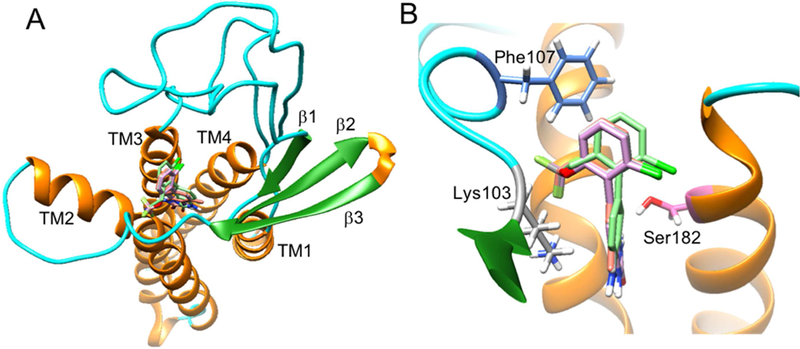
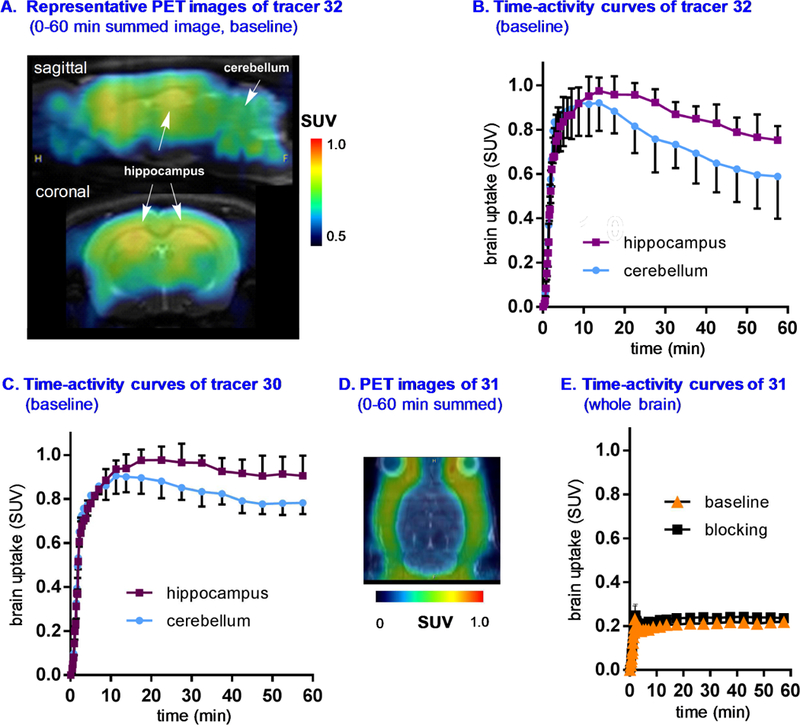
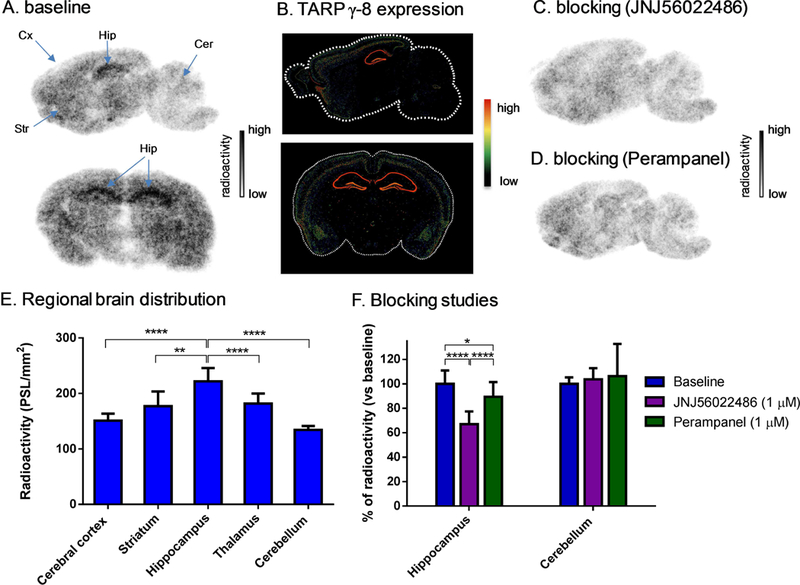
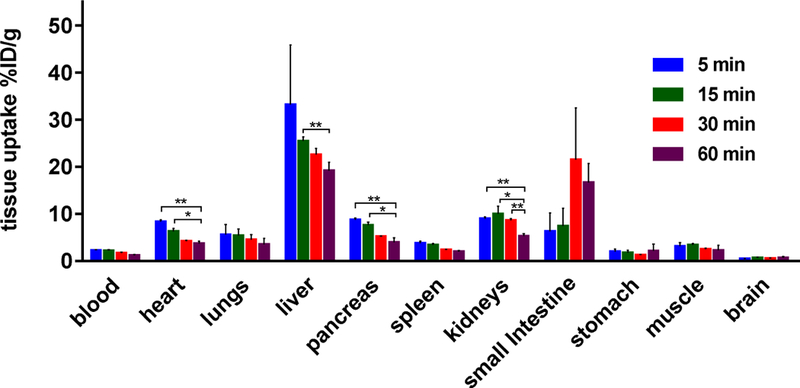
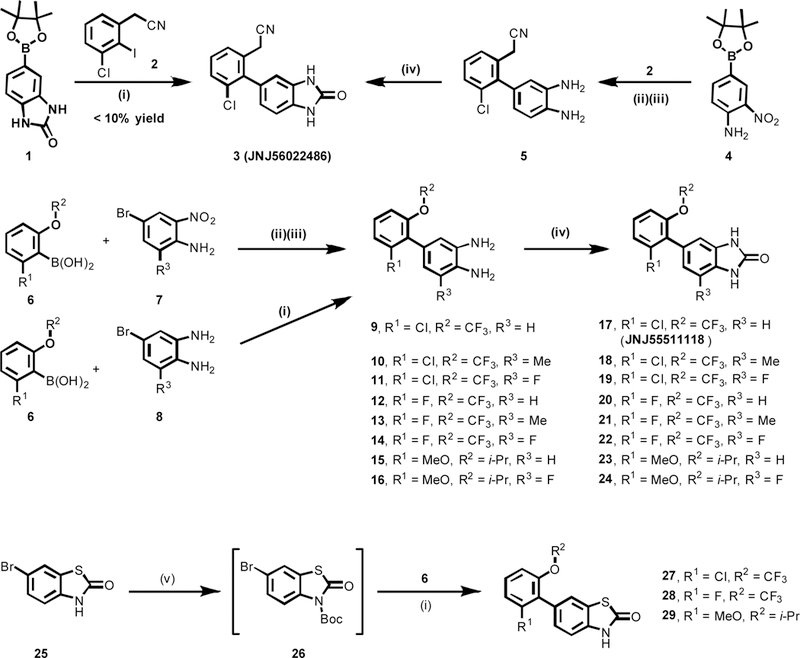
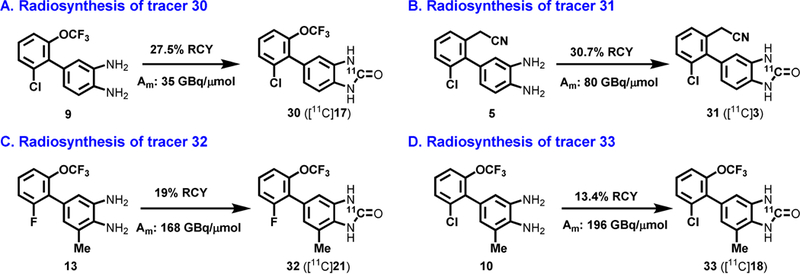
a-Amino-3-hydroxyl-5-methyl-4-isoxazolepropionic acid (AMPA) receptors are implicated in the pathology of neurological diseases such as epilepsy and schizophrenia. As pan antagonists for this target are often accompanied with undesired effects at high doses, one of the recent drug discovery approaches has shifted to subtype-selective AMPA receptor (AMPAR) antagonists, specifically, via modulating transmembrane AMPAR regulatory proteins (TARPs). The quantification of AMPARs by positron emission tomography (PET) would help obtain insights into disease conditions in the living brain and advance the translational development of AMPAR antagonists. Herein we report the design, synthesis and preclinical evaluation of a series of TARP γ-8 antagonists, amenable for radiolabeling, for the development of subtype-selective AMPAR PET imaging agents. Based on the pharmacology evaluation, molecular docking studies and physiochemical properties, we have identified several promising lead compounds 3, 17-19 and 21 for in vivo PET studies. All candidate compounds were labeled with [C]COCl in high radiochemical yields (13-31% RCY) and high molar activities (35-196 GBq/μmol). While tracers 30 ([C]17) &32 ([C]21) crossed the blood-brain barrier and showed heterogeneous distribution in PET studies, consistent with TARP γ-8 expression, high nonspecific binding prevented further evaluation. To our delight, tracer 31 ([C]3) showed good in vitro specific binding and characteristic high uptake in the hippocampus in rat brain tissues, which provides the guideline for further development of a new generation subtype selective TARP γ-8 dependent AMPAR tracers.
α-氨基-3-羟基-5-甲基-4-异恶唑丙酸(AMPA)受体与癫痫和精神分裂症等神经疾病的病理学有关。由于针对该靶点的泛拮抗剂在高剂量下常常伴随着不良作用,最近的药物发现方法之一已经转向 AMPA 受体(AMPAR)亚型选择性拮抗剂,特别是通过调节跨膜 AMPAR 调节蛋白(TARPs)。正电子发射断层扫描(PET)对 AMPAR 的定量将有助于深入了解活体大脑中的疾病状况,并推进 AMPAR 拮抗剂的转化发展。在此,我们报告了一系列 TARP γ-8 拮抗剂的设计、合成和临床前评估,这些拮抗剂可进行放射性标记,用于开发亚型选择性 AMPAR PET 成像剂。基于药理学评估、分子对接研究和物理化学性质,我们已经确定了几个有前途的候选化合物 3、17-19 和 21,用于体内 PET 研究。所有候选化合物均以高放射性化学产率(13-31%RCY)和高摩尔活性(35-196GBq/μmol)用 [C]COCl 标记。虽然示踪剂 30([C]17) 和 32([C]21) 穿过血脑屏障,在 PET 研究中表现出不均匀的分布,与 TARP γ-8 表达一致,但高非特异性结合阻止了进一步的评估。令我们高兴的是,示踪剂 31([C]3) 在体外表现出良好的特异性结合,并在大鼠脑组织中显示出特征性的高摄取量,这为进一步开发新一代依赖 TARP γ-8 的亚型选择性 AMPAR 示踪剂提供了指导。