University of Malawi, College of Medicine, Blantyre, Malawi
Malawi-Liverpool-Wellcome Trust Clinical Research Programme, Blantyre, Malawi.
Antimicrob Agents Chemother. 2018 Oct 24;62(11). doi: 10.1128/AAC.01162-18. Print 2018 Nov.
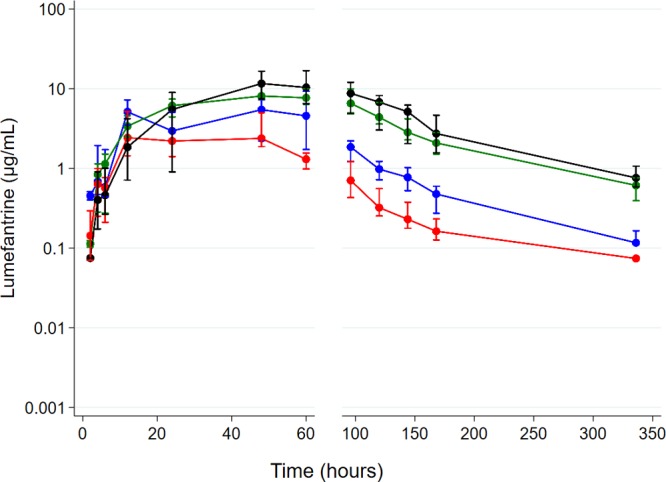
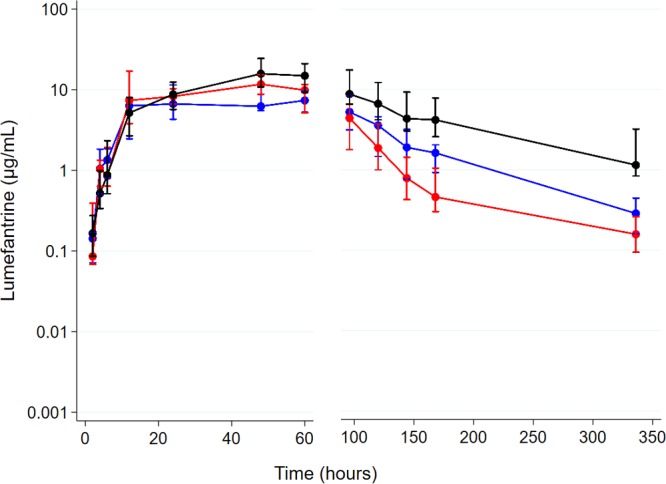
There is conflicting evidence of the impact of commonly used antiretroviral therapies (ARTs) on the pharmacokinetics of lumefantrine and the safety profile of artemether-lumefantrine. We compared the area under the concentration-time curve from 0 h to 14 days (AUC) of lumefantrine and the safety profile of artemether-lumefantrine in malaria-negative human immunodeficiency virus (HIV)-infected adults in two steps. In step 1, a half-dose adult course of artemether-lumefantrine was administered as a safety check in four groups ( = 6/group): (i) antiretroviral naive, (ii) nevirapine-based ART, (iii) efavirenz-based ART, and (iv) ritonavir-boosted lopinavir-based ART. In step 2, a standard-dose adult course of artemether-lumefantrine was administered to a different cohort in three groups ( = 10 to 15/group): (i) antiretroviral naive, (ii) efavirenz-based ART, and (iii) ritonavir-boosted lopinavir-based ART. In step 1, lumefantrine's AUC was 53% (95% confidence interval [CI], 0.27 to 0.82) lower in the efavirenz-based ART group than in the ART-naive group and was 2.4 (95% CI, 1.58 to 3.62) and 2.9(95% CI, 1.75 to 4.72) times higher in the nevirapine- and ritonavir-boosted lopinavir groups, respectively. In step 2, lumefantrine's AUC was 1.9 (95% CI, 1.26 to 3.00) times higher in the ritonavir-boosted lopinavir group and not significantly different between the efavirenz- and ART-naive groups (0.99 [95% CI, 0.63 to 1.57]). Frequent cases of hematological abnormalities (thrombocytopenia and neutropenia) were observed in the nevirapine group in step 1, leading to a recommendation from the data and safety monitoring board not to include a nevirapine group in step 2. Artemether-lumefantrine was well tolerated in the other groups. The therapeutic implications of these findings need to be evaluated among HIV-malaria-coinfected adults. (This study has been registered at the Pan African Clinical Trials Registry under numbers PACTR2010030001871293 and PACTR2010030001971409.).
抗逆转录病毒疗法对青蒿琥酯-咯萘啶药代动力学和安全性的影响存在相互矛盾的证据。我们分两步比较了疟疾阴性人类免疫缺陷病毒(HIV)感染成人中青蒿琥酯的浓度-时间曲线下面积(AUC)0 小时至 14 天(AUC)和青蒿琥酯-咯萘啶的安全性特征。在第 1 步中,我们在 4 组(每组 6 人)中给予青蒿琥酯-咯萘啶半剂量成人疗程作为安全性检查:(i)抗逆转录病毒治疗初治,(ii)奈韦拉平为基础的抗逆转录病毒治疗,(iii)依非韦伦为基础的抗逆转录病毒治疗,和(iv)利托那韦增效洛匹那韦为基础的抗逆转录病毒治疗。在第 2 步中,我们在 3 组(每组 10 至 15 人)中给予青蒿琥酯-咯萘啶标准剂量成人疗程:(i)抗逆转录病毒治疗初治,(ii)依非韦伦为基础的抗逆转录病毒治疗,和(iii)利托那韦增效洛匹那韦为基础的抗逆转录病毒治疗。在第 1 步中,依非韦伦为基础的抗逆转录病毒治疗组青蒿琥酯 AUC 比抗逆转录病毒治疗初治组低 53%(95%可信区间[CI],0.27 至 0.82),而奈韦拉平组和利托那韦增效洛匹那韦组分别高 2.4(95%CI,1.58 至 3.62)和 2.9(95%CI,1.75 至 4.72)倍。在第 2 步中,利托那韦增效洛匹那韦组青蒿琥酯 AUC 高 1.9 倍(95%CI,1.26 至 3.00),而依非韦伦组与抗逆转录病毒治疗初治组无显著差异(0.99 [95%CI,0.63 至 1.57])。第 1 步中奈韦拉平组频繁出现血液学异常(血小板减少和中性粒细胞减少),数据和安全监测委员会建议不再纳入奈韦拉平组,因此不纳入第 2 步。其他组青蒿琥酯-咯萘啶均耐受良好。这些发现的治疗意义需要在 HIV-疟疾合并感染成人中进行评估。(本研究已在泛非临床试验注册处注册,注册号为 PACTR2010030001871293 和 PACTR2010030001971409。)