Duygu Burcu, Poels Ella M, Juni Rio, Bitsch Nicole, Ottaviani Lara, Olieslagers Servé, de Windt Leon J, da Costa Martins Paula A
Department of Cardiology, CARIM School for Cardiovascular Diseases, Maastricht University, 6229 ER, Maastricht, The Netherlands.
Department of Physiology and Cardiothoracic Surgery, Faculty of Medicine, University of Porto, 4099-002, Porto, Portugal.
Noncoding RNA Res. 2017 Jan 13;2(1):18-26. doi: 10.1016/j.ncrna.2016.12.002. eCollection 2017 Mar.
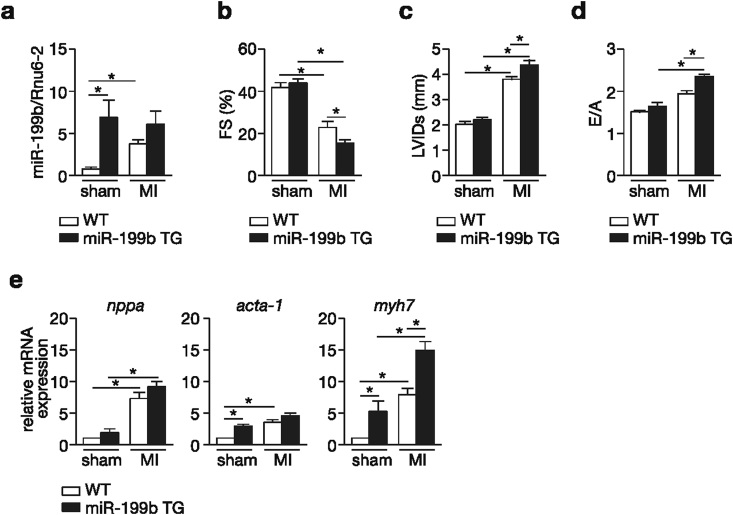
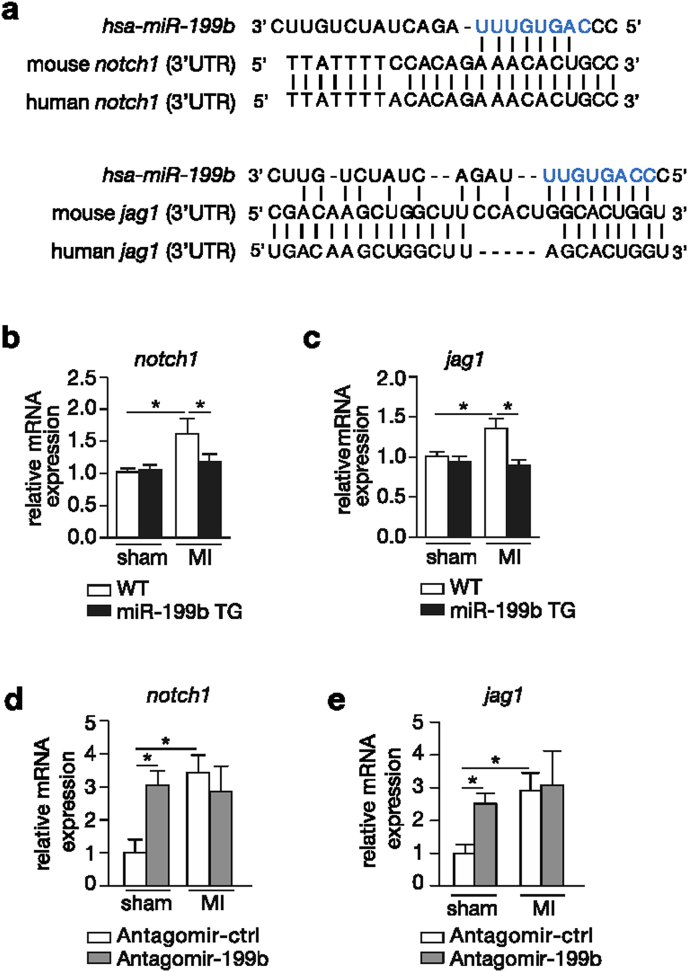
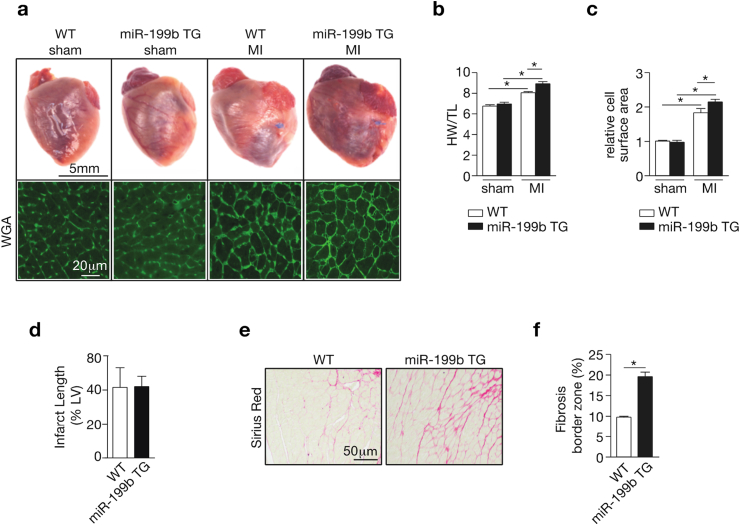
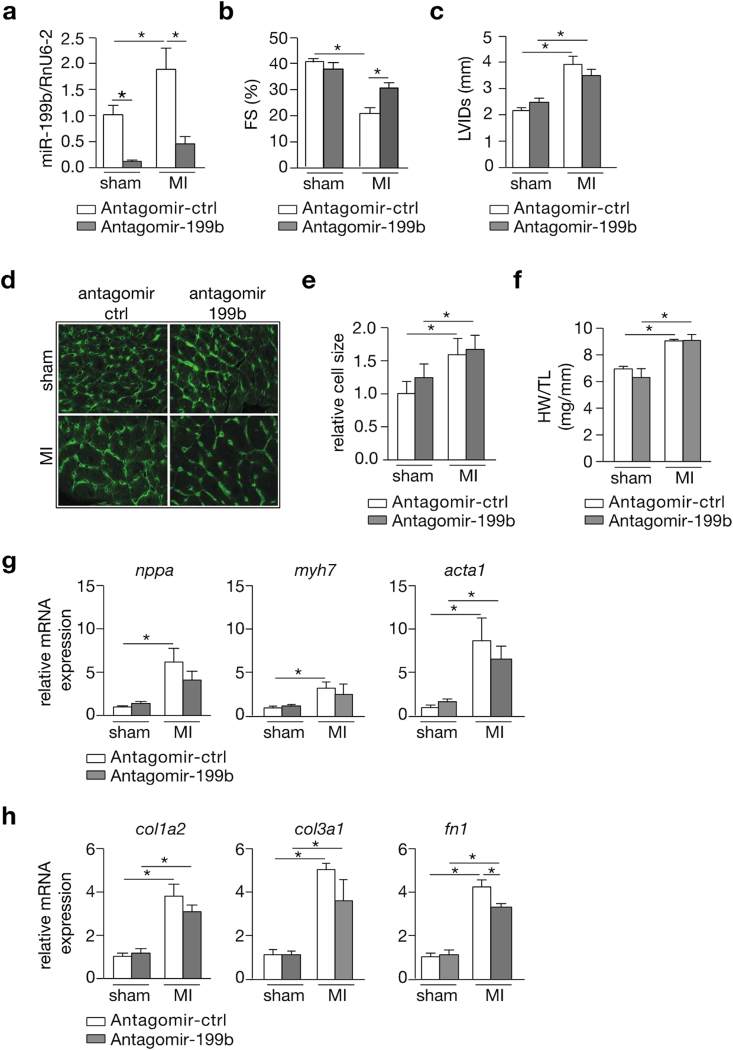
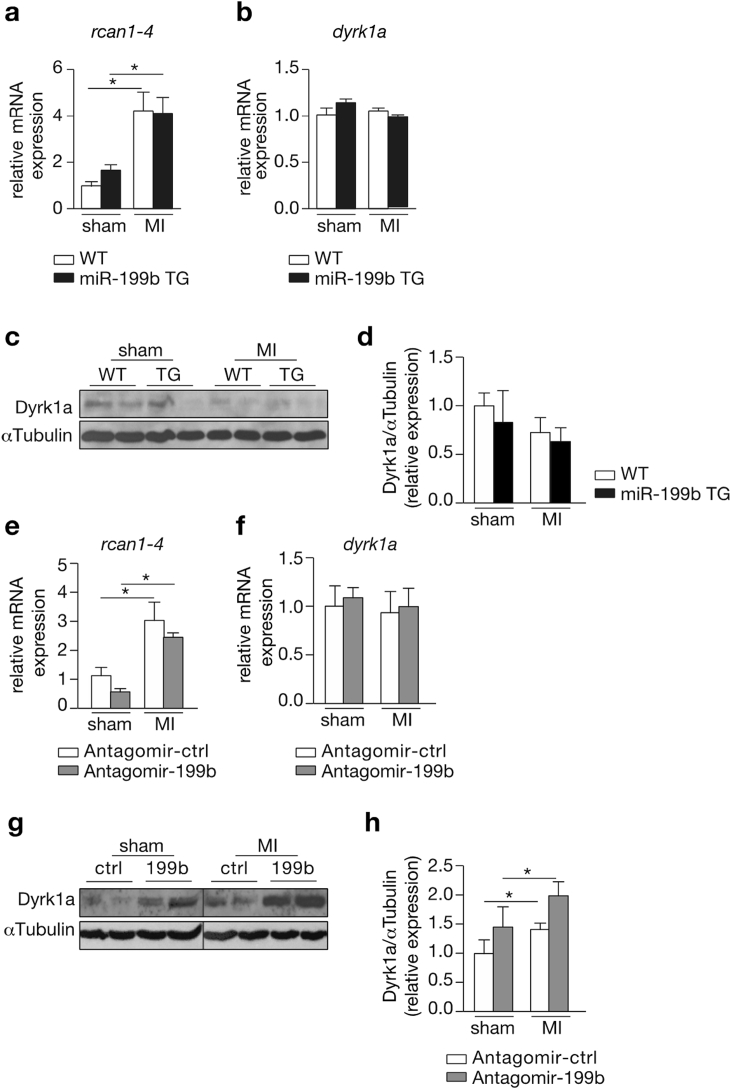
Myocardial infarction (MI), the globally leading cause of heart failure, morbidity and mortality, involves post-MI ventricular remodeling, a complex process including acute injury healing, scar formation and global changes in the surviving myocardium. The molecular mechanisms involved in adverse post-infarct left ventricular remodeling still remain poorly defined. Recently, microRNAs have been implicated in the development and progression of various cardiac diseases as crucial regulators of gene expression. We previously demonstrated that in a murine model of pressure overload, a model of heart failure secondary to aortic stenosis or chronic high blood pressure, elevated myocardial expression of miR-199b-5p is sufficient to activate calcineurin/NFAT signaling, leading to exaggerated cardiac pathological remodeling and dysfunction. Given the differences in left ventricular remodeling secondary to post-infarct healing and pressure overload, we evaluated miR-199b function in post-MI remodeling. We confirmed that the expression of miR-199b is elevated in the post-infarcted heart. Transgenic animals with cardiomyocyte-restricted overexpression of miR-199b-5p displayed exaggerated pathological remodeling after MI, reflected by severe systolic and diastolic dysfunction and fibrosis deposition. Conversely, therapeutic silencing of miR-199b-5p in MI-induced cardiac remodeling by using an antagomir to specifically inhibit endogenous miR-199b-5p , resulted in efficient suppression of cardiac miR-199b-5p expression and attenuated cardiac dysfunction and dilation following MI. Mechanistically, miR-199b-5p influenced the expression of three predicted target genes in post-infarcted hearts, dual specificity tyrosine-phosphorylation-regulated kinase 1A (Dyrk1a), the notch1 receptor and its ligand jagged1. In conclusion, here we provide evidence supporting that stress-induced miR-199b-5p participates in post-infarct remodeling by simultaneous regulation of distinct target genes.
心肌梗死(MI)是全球范围内导致心力衰竭、发病和死亡的主要原因,涉及心肌梗死后心室重塑,这是一个复杂的过程,包括急性损伤愈合、瘢痕形成以及存活心肌的整体变化。心肌梗死后左心室不良重塑所涉及的分子机制仍不清楚。最近,微小RNA作为基因表达的关键调节因子,已被证明与各种心脏疾病的发生和发展有关。我们之前证明,在压力超负荷的小鼠模型中,即主动脉狭窄或慢性高血压继发的心力衰竭模型中,心肌中miR-199b-5p表达升高足以激活钙调神经磷酸酶/活化T细胞核因子信号通路,导致心脏病理重塑和功能障碍加剧。鉴于心肌梗死后愈合和压力超负荷继发的左心室重塑存在差异,我们评估了miR-199b在心肌梗死后重塑中的作用。我们证实,心肌梗死后心脏中miR-199b的表达升高。心肌细胞特异性过表达miR-199b-5p的转基因动物在心肌梗死后表现出过度的病理重塑,表现为严重的收缩和舒张功能障碍以及纤维化沉积。相反,通过使用抗miR来特异性抑制内源性miR-199b-5p,对心肌梗死诱导的心脏重塑进行治疗性沉默,可有效抑制心脏miR-199b-5p的表达,并减轻心肌梗死后的心脏功能障碍和扩张。从机制上讲,miR-199b-5p影响心肌梗死后心脏中三个预测靶基因的表达,即双特异性酪氨酸磷酸化调节激酶1A(Dyrk1a)、Notch1受体及其配体锯齿蛋白1。总之,我们提供的证据支持应激诱导的miR-199b-5p通过同时调节不同的靶基因参与心肌梗死后的重塑。