Department of Biosystems Science and Engineering, ETH Zürich, Basel, Switzerland
Swiss Institute of Bioinformatics (SIB), Switzerland.
J R Soc Interface. 2018 Sep;15(146). doi: 10.1098/rsif.2018.0512.
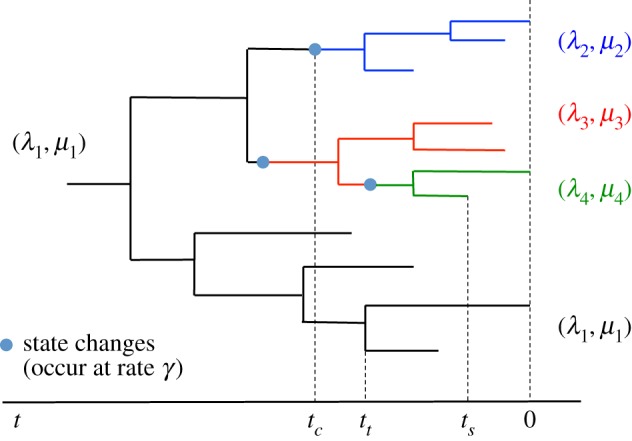
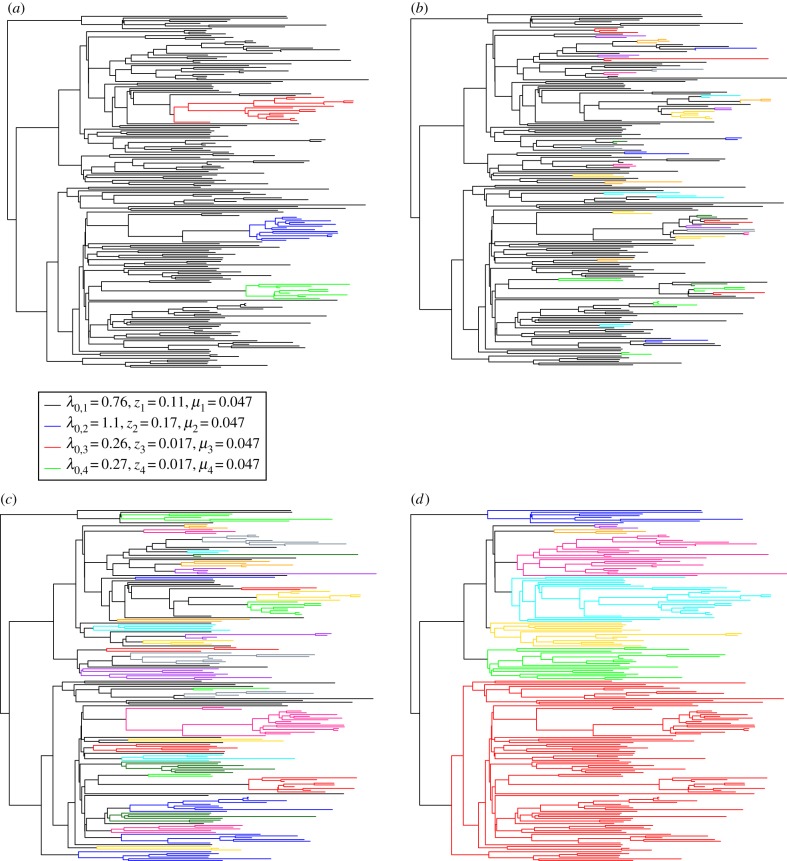
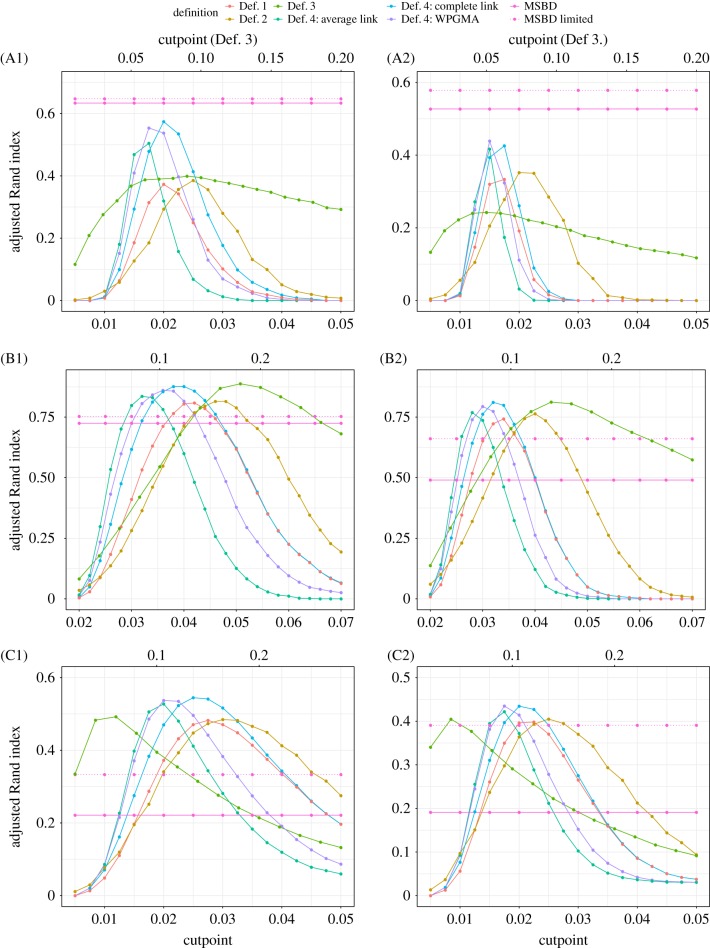
HIV patients form clusters in HIV transmission networks. Accurate identification of these transmission clusters is essential to effectively target public health interventions. One reason for clustering is that the underlying contact network contains many local communities. We present a new maximum-likelihood method for identifying transmission clusters caused by community structure, based on phylogenetic trees. The method employs a multi-state birth-death (MSBD) model which detects changes in transmission rate, which are interpreted as the introduction of the epidemic into a new susceptible community, i.e. the formation of a new cluster. We show that the MSBD method is able to reliably infer the clusters and the transmission parameters from a pathogen phylogeny based on our simulations. In contrast to existing cutpoint-based methods for cluster identification, our method does not require that clusters be monophyletic nor is it dependent on the selection of a difficult-to-interpret cutpoint parameter. We present an application of our method to data from the Swiss HIV Cohort Study. The method is available as an easy-to-use R package.
HIV 患者在 HIV 传播网络中形成簇。准确识别这些传播簇对于有效针对公共卫生干预措施至关重要。形成簇的一个原因是,潜在的接触网络包含许多局部社区。我们提出了一种新的基于系统发育树识别社区结构引起的传播簇的最大似然方法。该方法采用多状态 Birth-Death (MSBD) 模型来检测传播率的变化,这些变化被解释为传染病进入新的易感社区,即形成新的簇。我们通过模拟表明,MSBD 方法能够可靠地从病原体系统发育推断出簇和传播参数。与现有的基于切点的聚类识别方法不同,我们的方法不需要聚类是单系的,也不依赖于选择难以解释的切点参数。我们提出了一种应用我们的方法的应用,用于来自瑞士 HIV 队列研究的数据。该方法可用作易于使用的 R 包。