Giardina Federica, Romero-Severson Ethan Obie, Albert Jan, Britton Tom, Leitner Thomas
Department of Mathematics, Stockholm University, Stockholm, Sweden.
Theoretical Biology and Biophysics Group, Los Alamos National Laboratory, Los Alamos, New Mexico, United States of America.
PLoS Comput Biol. 2017 Jan 13;13(1):e1005316. doi: 10.1371/journal.pcbi.1005316. eCollection 2017 Jan.
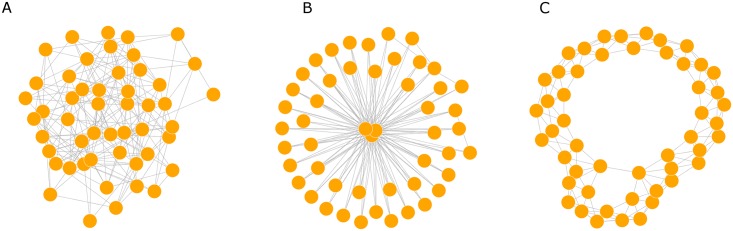
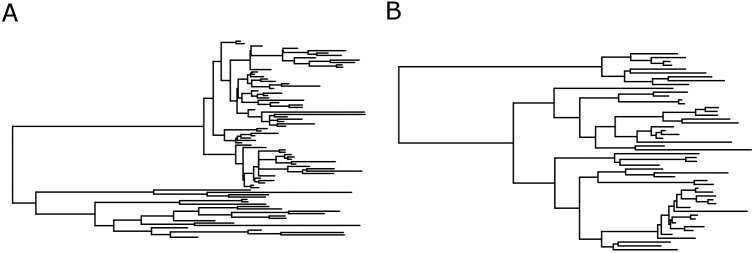
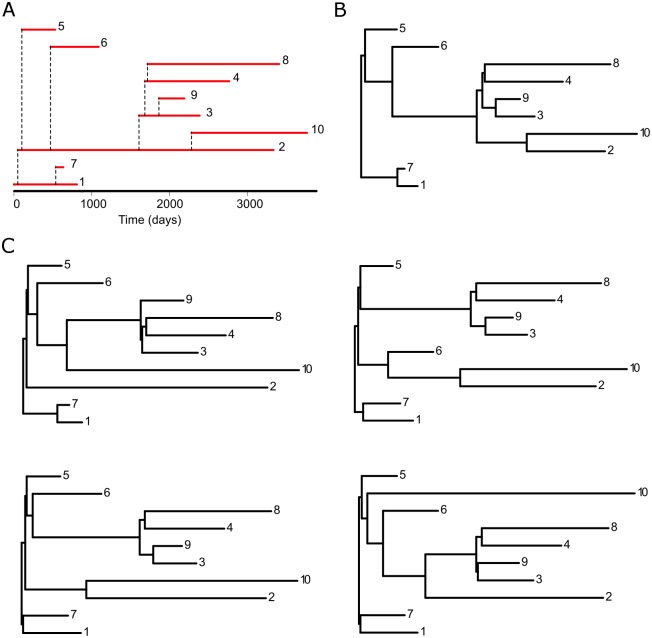
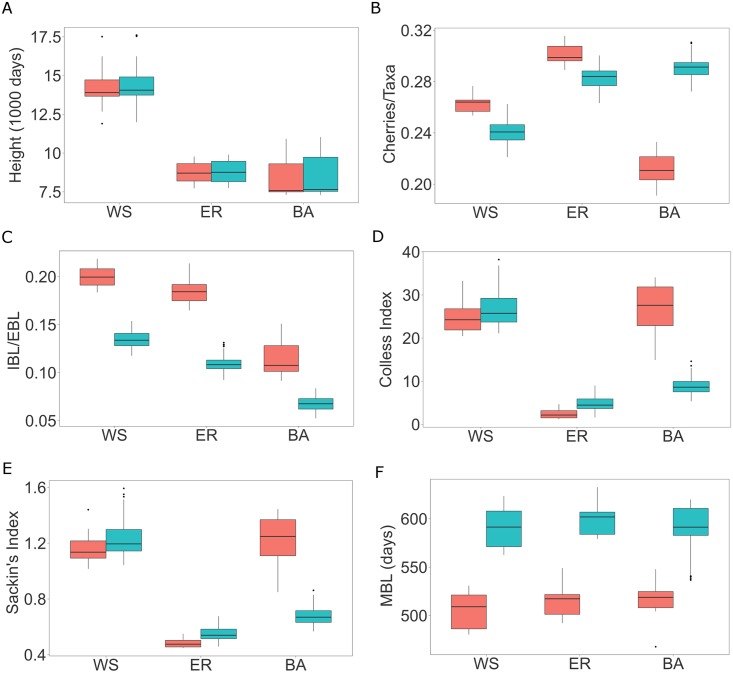
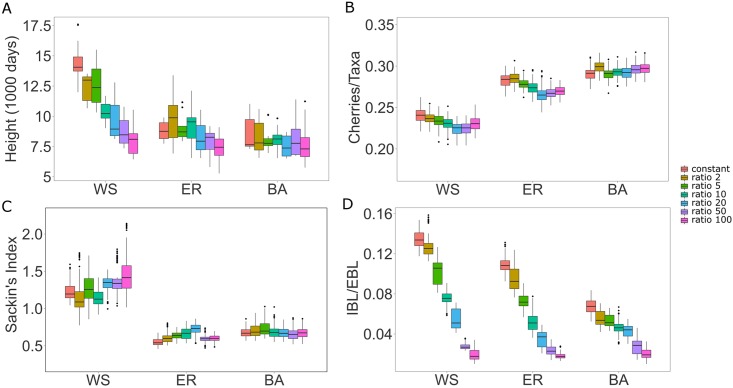
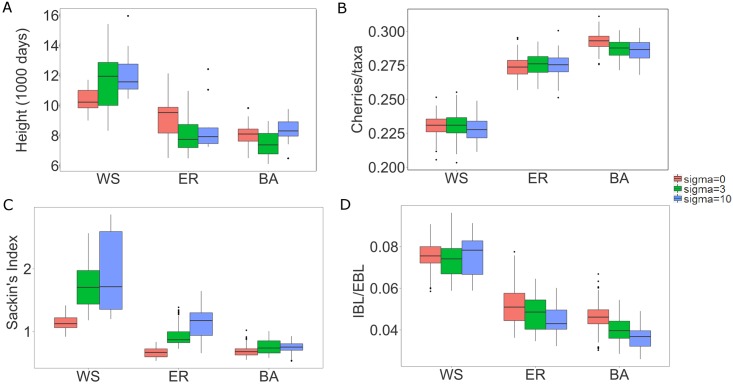
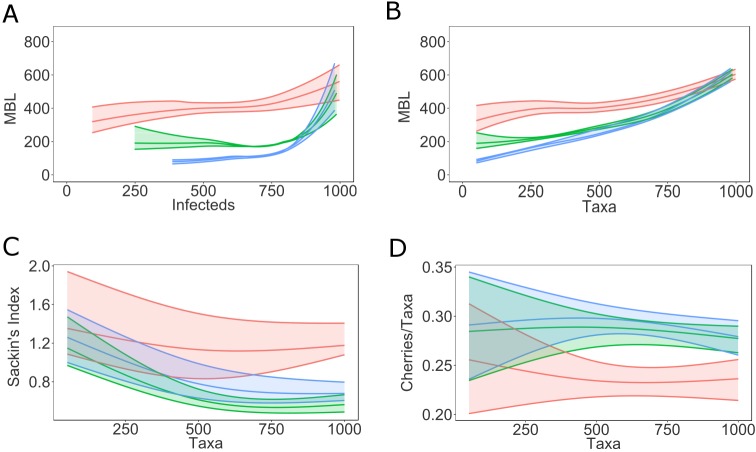
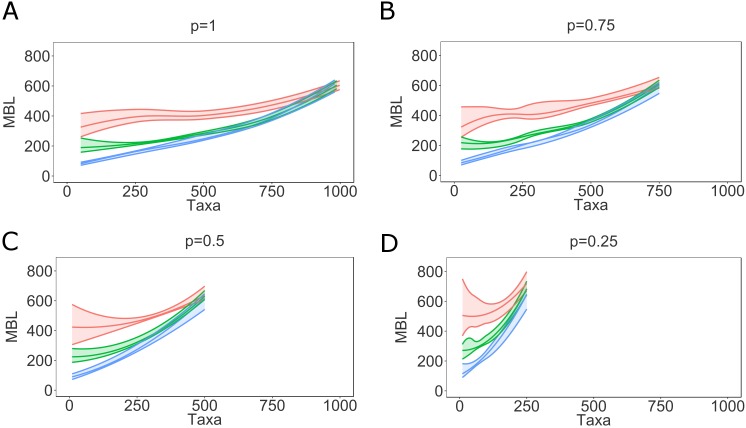
Phylogenetic inference is an attractive means to reconstruct transmission histories and epidemics. However, there is not a perfect correspondence between transmission history and virus phylogeny. Both node height and topological differences may occur, depending on the interaction between within-host evolutionary dynamics and between-host transmission patterns. To investigate these interactions, we added a within-host evolutionary model in epidemiological simulations and examined if the resulting phylogeny could recover different types of contact networks. To further improve realism, we also introduced patient-specific differences in infectivity across disease stages, and on the epidemic level we considered incomplete sampling and the age of the epidemic. Second, we implemented an inference method based on approximate Bayesian computation (ABC) to discriminate among three well-studied network models and jointly estimate both network parameters and key epidemiological quantities such as the infection rate. Our ABC framework used both topological and distance-based tree statistics for comparison between simulated and observed trees. Overall, our simulations showed that a virus time-scaled phylogeny (genealogy) may be substantially different from the between-host transmission tree. This has important implications for the interpretation of what a phylogeny reveals about the underlying epidemic contact network. In particular, we found that while the within-host evolutionary process obscures the transmission tree, the diversification process and infectivity dynamics also add discriminatory power to differentiate between different types of contact networks. We also found that the possibility to differentiate contact networks depends on how far an epidemic has progressed, where distance-based tree statistics have more power early in an epidemic. Finally, we applied our ABC inference on two different outbreaks from the Swedish HIV-1 epidemic.
系统发育推断是重建传播历史和疫情的一种有吸引力的方法。然而,传播历史与病毒系统发育之间并不存在完美的对应关系。节点高度和拓扑差异都可能出现,这取决于宿主内进化动态与宿主间传播模式之间的相互作用。为了研究这些相互作用,我们在流行病学模拟中加入了宿主内进化模型,并检验由此产生的系统发育能否恢复不同类型的接触网络。为了进一步提高真实性,我们还引入了疾病各阶段传染性的患者特异性差异,并且在疫情层面考虑了不完全抽样和疫情的持续时间。其次,我们实施了一种基于近似贝叶斯计算(ABC)的推断方法,以区分三种经过充分研究的网络模型,并联合估计网络参数和关键流行病学数量,如感染率。我们的ABC框架使用基于拓扑和距离的树统计量来比较模拟树和观察树。总体而言,我们的模拟表明,病毒时间尺度系统发育(谱系)可能与宿主间传播树有很大不同。这对于解释系统发育所揭示的潜在疫情接触网络具有重要意义。特别是,我们发现虽然宿主内进化过程模糊了传播树,但多样化过程和传染性动态也增加了区分不同类型接触网络的鉴别力。我们还发现区分接触网络的可能性取决于疫情发展的程度,其中基于距离的树统计量在疫情早期更具鉴别力。最后,我们将ABC推断应用于瑞典HIV-1疫情的两次不同爆发。