Department of Medicine, Indiana University School of Medicine Indianapolis, Indianapolis, IN, 46202, USA.
Department of Hematology and Oncology, Taussig Cancer Center, Cleveland Clinic, Cleveland, OH, 44195, USA.
Cell Death Dis. 2018 Sep 6;9(9):912. doi: 10.1038/s41419-018-0919-9.
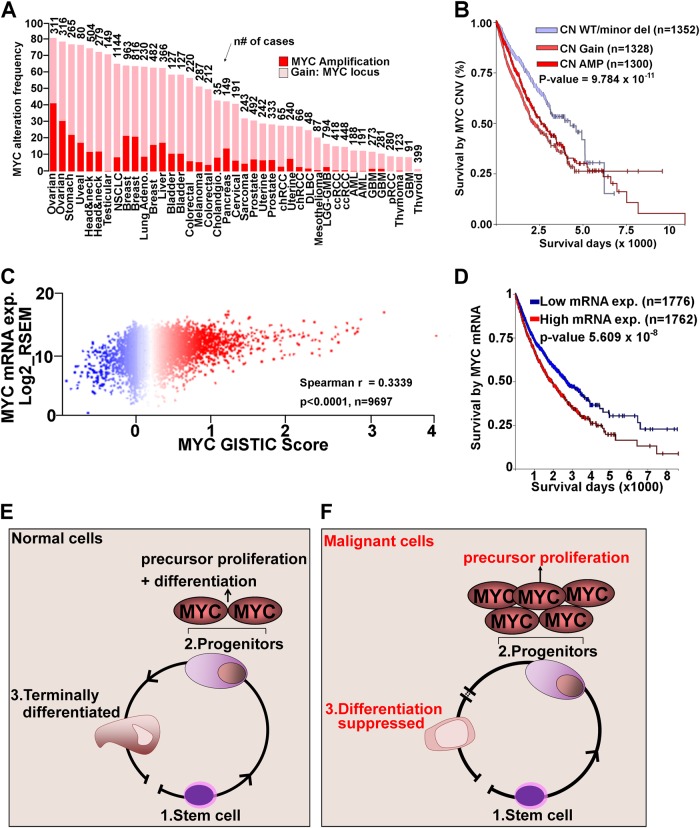
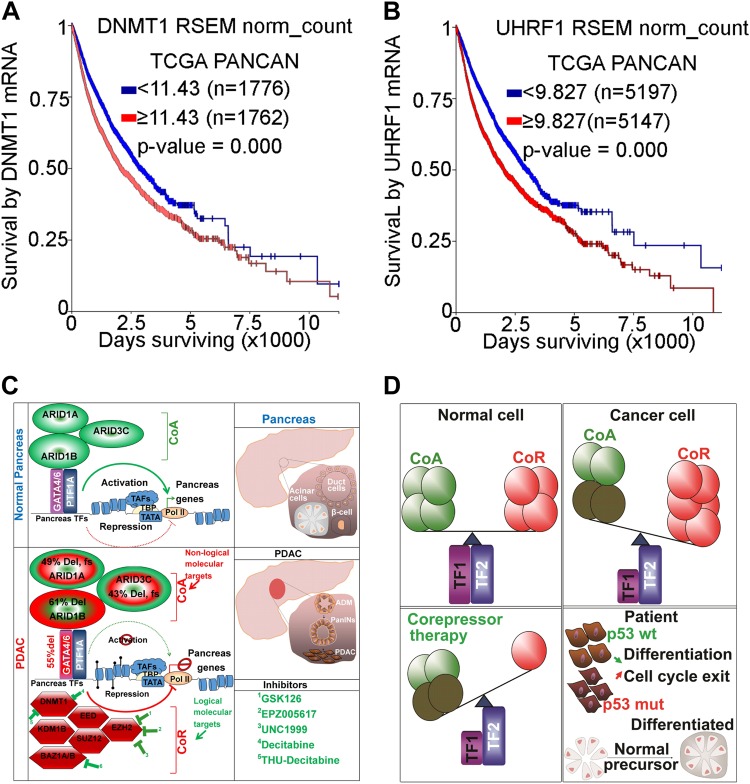
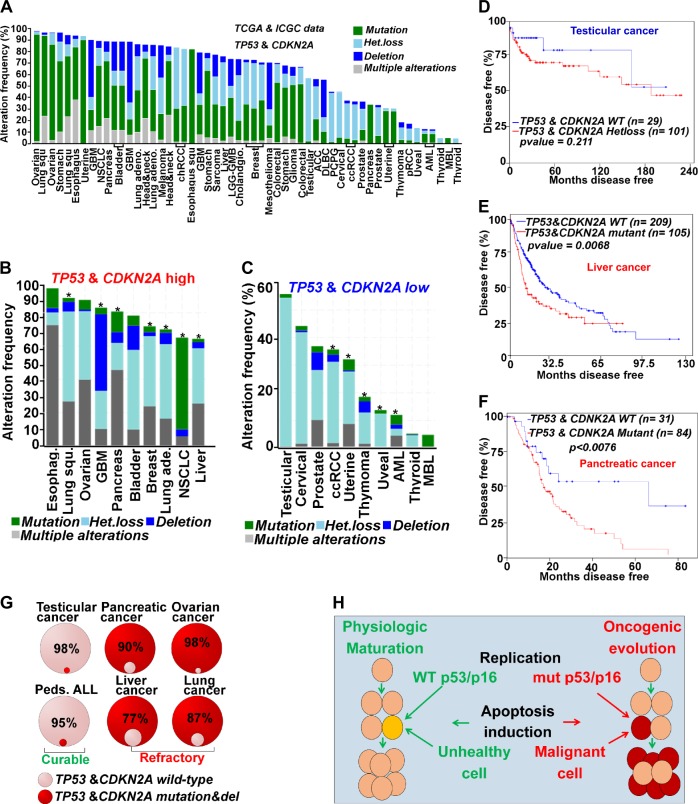
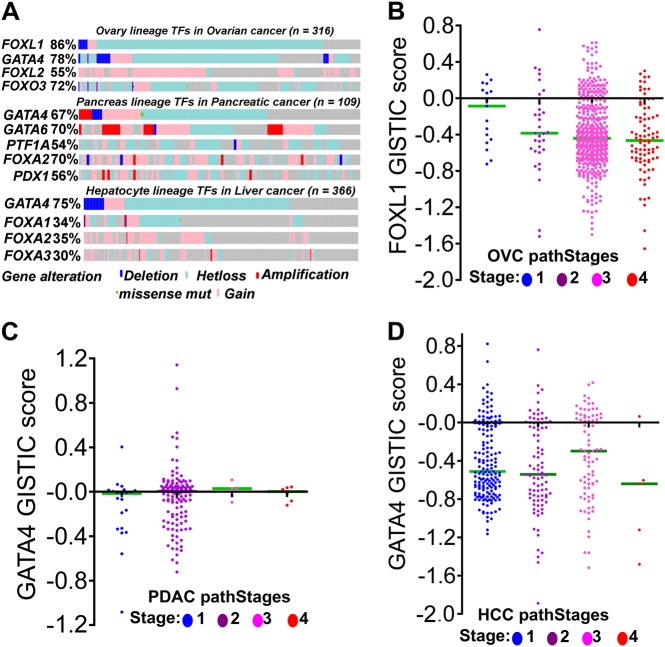
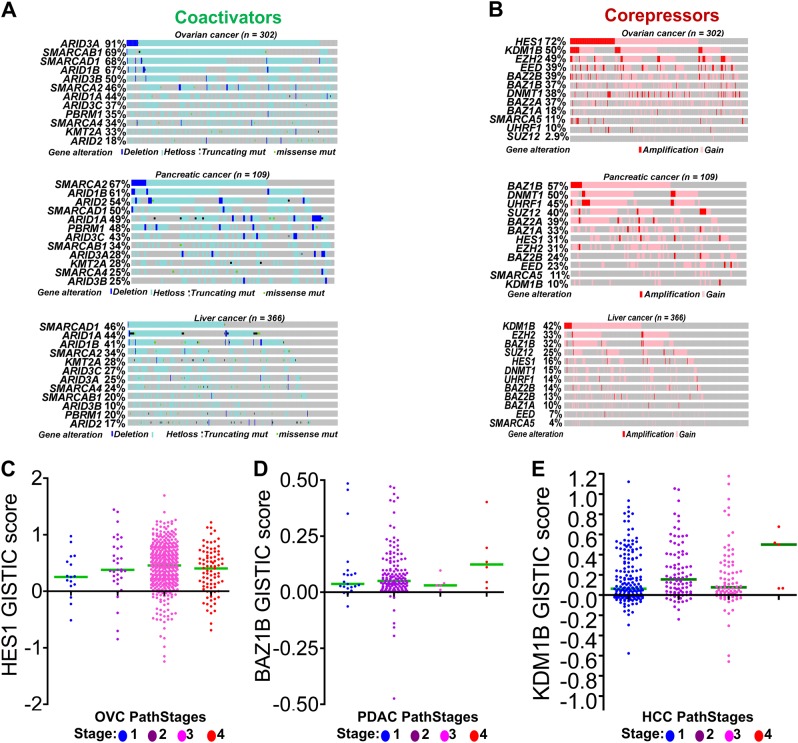
Chemotherapeutic drugs have a common intent to activate apoptosis in tumor cells. However, master regulators of apoptosis (e.g., p53, p16/CDKN2A) are frequently genetically inactivated in cancers, resulting in multidrug resistance. An alternative, p53-independent method for terminating malignant proliferation is to engage terminal-differentiation. Normally, the exponential proliferation of lineage-committed progenitors, coordinated by the master transcription factor (TF) MYC, is self-limited by forward-differentiation to terminal lineage-fates. In cancers, however, this exponential proliferation is disengaged from terminal-differentiation. The mechanisms underlying this decoupling are mostly unknown. We performed a systematic review of published literature (January 2007-June 2018) to identify gene pathways linked to differentiation-failure in three treatment-recalcitrant cancers: hepatocellular carcinoma (HCC), ovarian cancer (OVC), and pancreatic ductal adenocarcinoma (PDAC). We analyzed key gene alterations in various apoptosis, proliferation and differentiation pathways to determine whether it is possible to predict treatment outcomes and suggest novel therapies. Poorly differentiated tumors were linked to poorer survival across histologies. Our analyses suggested loss-of-function events to master TF drivers of lineage-fates and their cofactors as being linked to differentiation-failure: genomic data in TCGA and ICGC databases demonstrated frequent haploinsufficiency of lineage master TFs (e.g., GATA4/6) in poorly differentiated tumors; the coactivators that these TFs use to activate genes (e.g. ARID1A, PBRM1) were also frequently inactivated by genetic mutation and/or deletion. By contrast, corepressor components (e.g., DNMT1, EED, UHRF1, and BAZ1A/B), that oppose coactivators to repress or turn off genes, were frequently amplified instead, and the level of amplification was highest in poorly differentiated lesions. This selection by neoplastic evolution towards unbalanced activity of transcriptional corepressors suggests these enzymes as candidate targets for inhibition aiming to re-engage forward-differentiation. This notion is supported by both pre-clinical and clinical trial literature.
化疗药物的共同目的是激活肿瘤细胞中的细胞凋亡。然而,凋亡的主要调节因子(例如,p53、p16/CDKN2A)在癌症中经常发生遗传失活,导致多药耐药。另一种终止恶性增殖的方法是诱导终末分化。通常情况下,由主转录因子(TF)MYC 协调的谱系定向祖细胞的指数增殖受到向前分化为终末谱系命运的限制。然而,在癌症中,这种指数增殖与终末分化脱耦。这种脱耦的机制在很大程度上尚不清楚。我们对已发表的文献(2007 年 1 月至 2018 年 6 月)进行了系统回顾,以确定与三种治疗抵抗性癌症(肝细胞癌(HCC)、卵巢癌(OVC)和胰腺导管腺癌(PDAC))中的分化失败相关的基因途径。我们分析了各种凋亡、增殖和分化途径中的关键基因改变,以确定是否可以预测治疗结果并提出新的治疗方法。低分化肿瘤与不同组织学类型的生存率较差相关。我们的分析表明,谱系命运的主 TF 及其共因子的功能丧失事件与分化失败有关:TCGA 和 ICGC 数据库中的基因组数据表明,低分化肿瘤中谱系主 TF(例如,GATA4/6)的杂合子缺失频率较高;这些 TF 用于激活基因的共激活因子(例如,ARID1A、PBRM1)也经常因遗传突变和/或缺失而失活。相比之下,核心抑制因子成分(例如,DNMT1、EED、UHRF1 和 BAZ1A/B),它们与共激活因子相反,抑制或关闭基因,反而经常被扩增,在低分化病变中扩增程度最高。肿瘤进化对转录核心抑制因子的不平衡活性的这种选择表明这些酶是抑制它们以重新激活向前分化的候选靶标。这一观点得到了临床前和临床试验文献的支持。