Department of Periodontics, University of Washington, Box 357444, Seattle, WA, 98195, USA.
City of Hope, Information Sciences - Beckman Research Institute, 4920 Rivergrade Rd., Irwindale, CA, 91706, USA.
BMC Genomics. 2018 Sep 14;19(1):675. doi: 10.1186/s12864-018-5042-x.
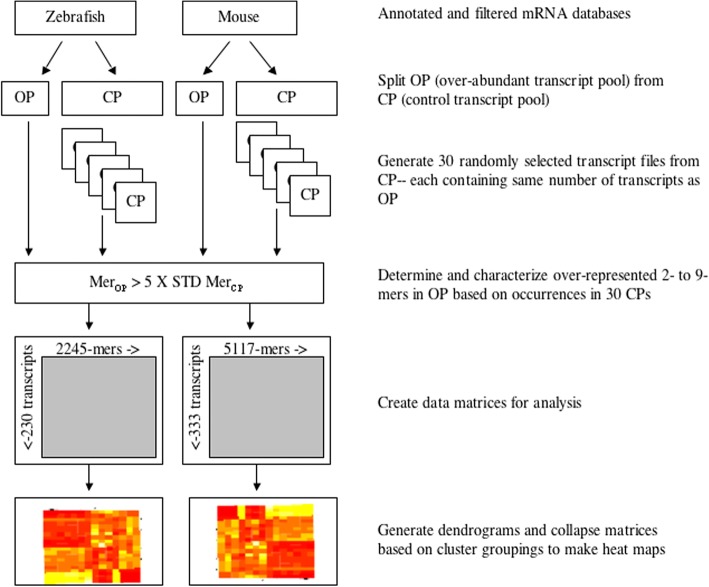
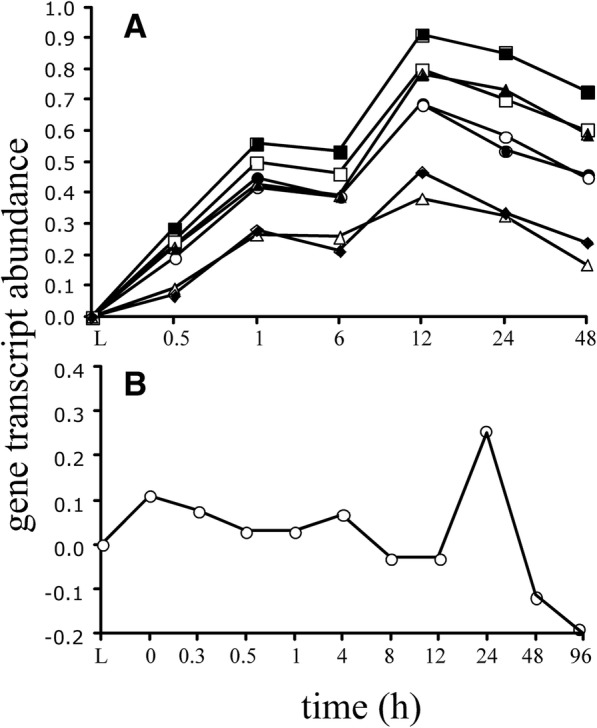
Our previous study found that more than 500 transcripts significantly increased in abundance in the zebrafish and mouse several hours to days postmortem relative to live controls. The current literature suggests that most mRNAs are post-transcriptionally regulated in stressful conditions. We rationalized that the postmortem transcripts must contain sequence features (3- to 9- mers) that are unique from those in the rest of the transcriptome and that these features putatively serve as binding sites for proteins and/or non-coding RNAs involved in post-transcriptional regulation.
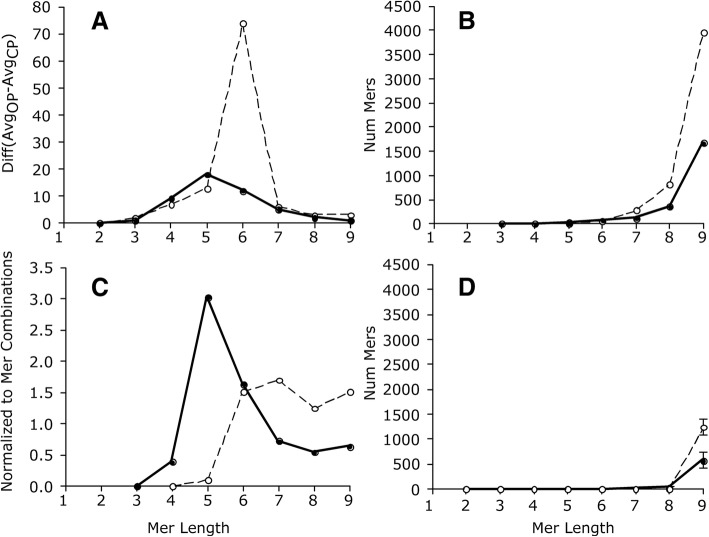
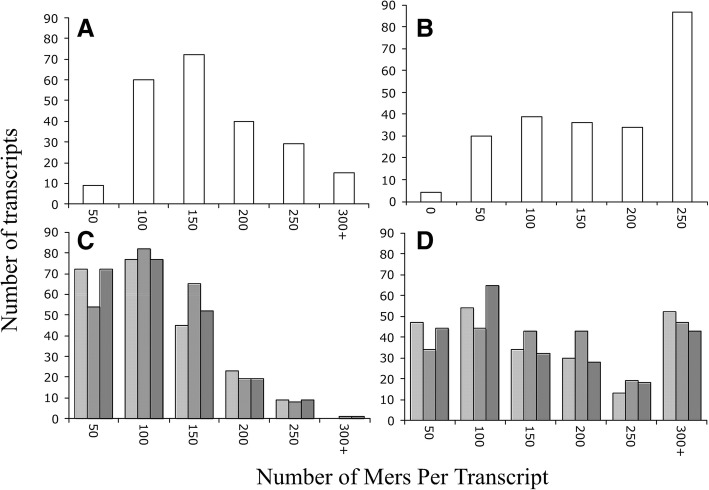
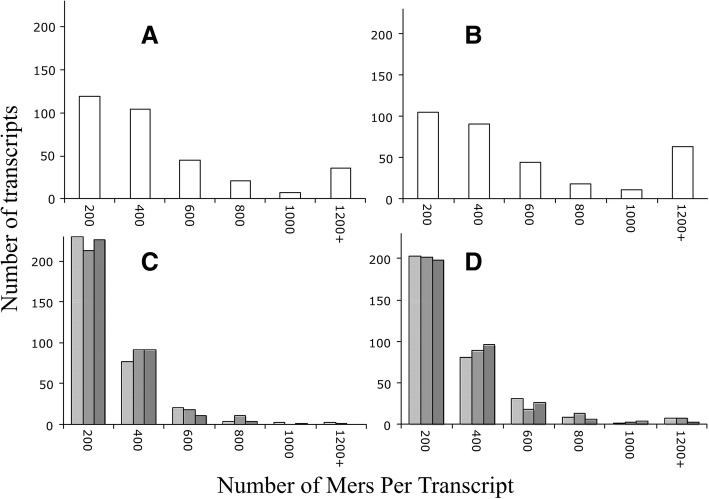
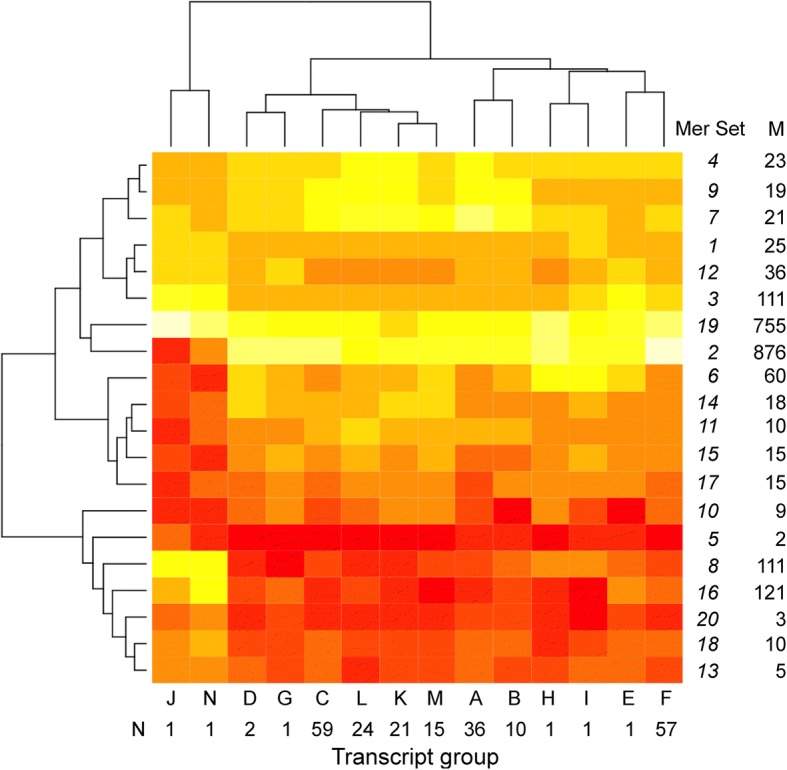
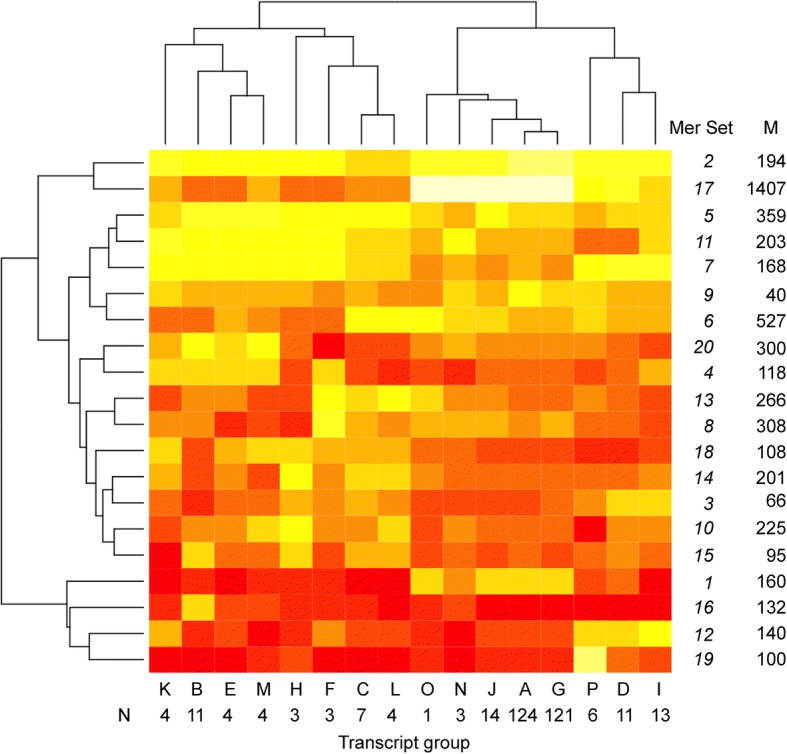
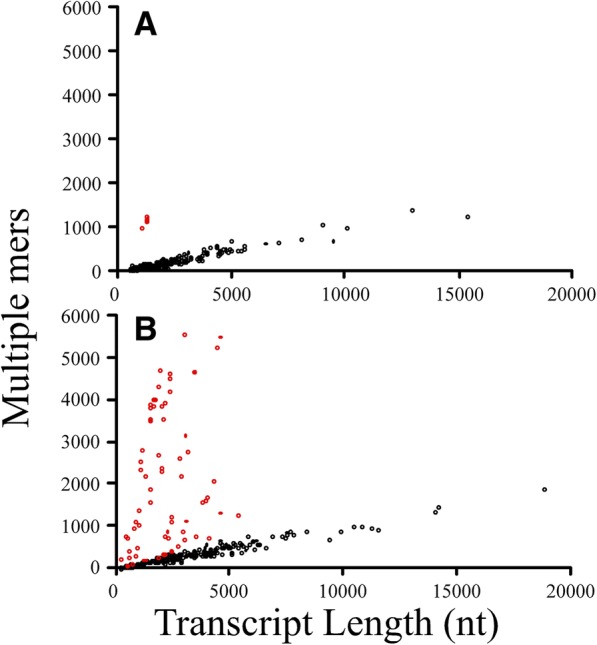
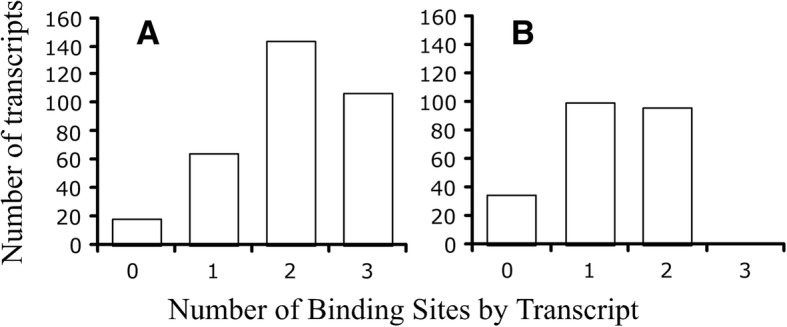
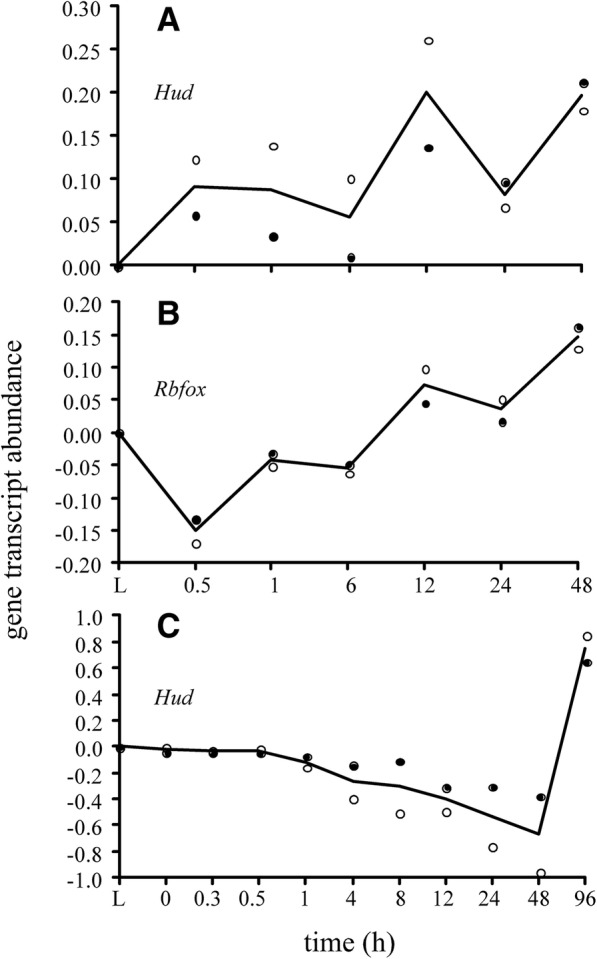
We identified 5117 and 2245 over-represented sequence features in the mouse and zebrafish, respectively, which represents less than 1.5% of all possible features. Some of these features were disproportionately distributed along the transcripts with high densities in the 3' untranslated regions of the zebrafish (0.3 mers/nt) and the open reading frames of the mouse (0.6 mers/nt). Yet, the highest density (2.3 mers/nt) occurred in the open reading frames of 11 mouse transcripts that lacked 3' or 5' untranslated regions. These results suggest the transcripts with high density of features might serve as 'molecular sponges' that sequester RNA binding proteins and/or microRNAs, and thus indirectly increase the stability and gene expression of other transcripts. In addition, some of the features were identified as binding sites for Rbfox and Hud proteins that are also involved in increasing transcript stability and gene expression.
Our results are consistent with the hypothesis that transcripts involved in responding to extreme stress, such as organismal death, have sequence features that make them different from the rest of the transcriptome. Some of these features serve as putative binding sites for proteins and non-coding RNAs that determine transcript stability and fate. A small number of the transcripts have high density sequence features, which are presumably involved in sequestering RNA binding proteins and microRNAs and thus preventing regulatory interactions among other transcripts. Our results provide baseline data on post-transcriptional regulation in stressful conditions that has implications for regulation in disease, starvation, and cancer.
我们之前的研究发现,与活体对照相比,死后数小时至数天的斑马鱼和小鼠中,有超过 500 个转录本的丰度显著增加。目前的文献表明,大多数 mRNA 在应激条件下是转录后调控的。我们推断,死后的转录本必须包含与转录组其余部分不同的序列特征(3-9 个核苷酸),这些特征推测可作为参与转录后调控的蛋白质和/或非编码 RNA 的结合位点。
我们分别在小鼠和斑马鱼中鉴定出 5117 和 2245 个过度表达的序列特征,它们不到所有可能特征的 1.5%。其中一些特征在转录本中分布不均,斑马鱼的 3'非翻译区(0.3 个核苷酸/nt)和小鼠的开放阅读框(0.6 个核苷酸/nt)高密度分布。然而,密度最高(2.3 个核苷酸/nt)的是 11 个缺乏 3'或 5'非翻译区的小鼠转录本的开放阅读框。这些结果表明,特征密度高的转录本可能充当“分子海绵”,隔离 RNA 结合蛋白和/或 microRNAs,从而间接增加其他转录本的稳定性和基因表达。此外,一些特征被鉴定为 Rbfox 和 Hud 蛋白的结合位点,它们也参与增加转录本的稳定性和基因表达。
我们的结果与以下假设一致,即参与应对极端应激(如机体死亡)的转录本具有使其与转录组其余部分不同的序列特征。其中一些特征可作为决定转录本稳定性和命运的蛋白质和非编码 RNA 的推定结合位点。少数转录本具有高密度的序列特征,可能参与隔离 RNA 结合蛋白和 microRNAs,从而防止其他转录本之间的调节相互作用。我们的结果提供了应激条件下转录后调控的基线数据,这对疾病、饥饿和癌症中的调控具有重要意义。