Department of Computational Medicine & Bioinformatics, University of Michigan, Ann Arbor, MI, 48109, USA.
McDonnell Genome Institute & Department of Medicine, Washington University, St. Louis, MO, 63108, USA.
Nat Commun. 2018 Sep 14;9(1):3753. doi: 10.1038/s41467-018-05936-5.
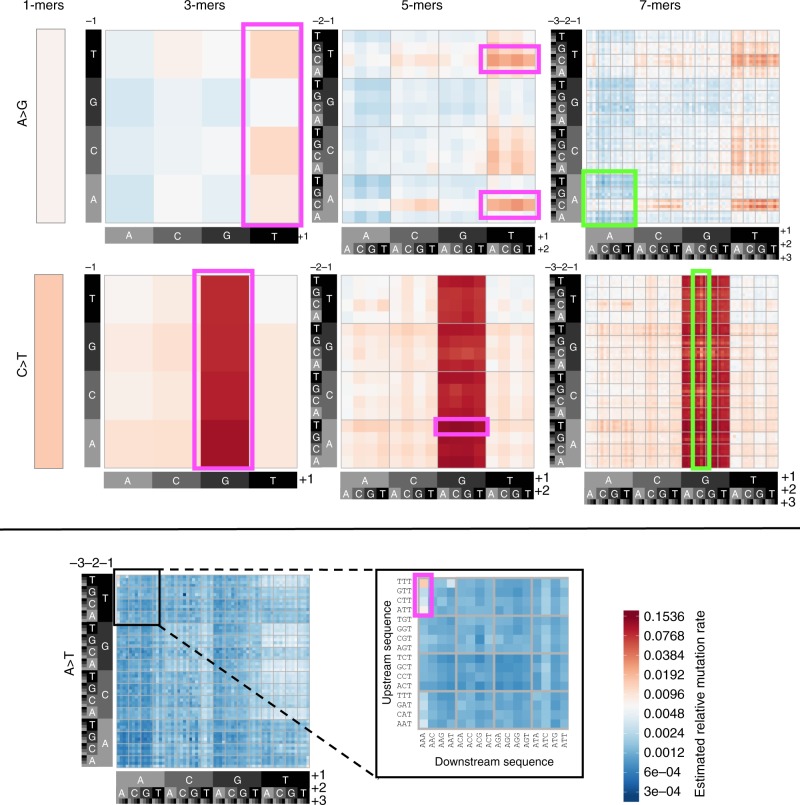
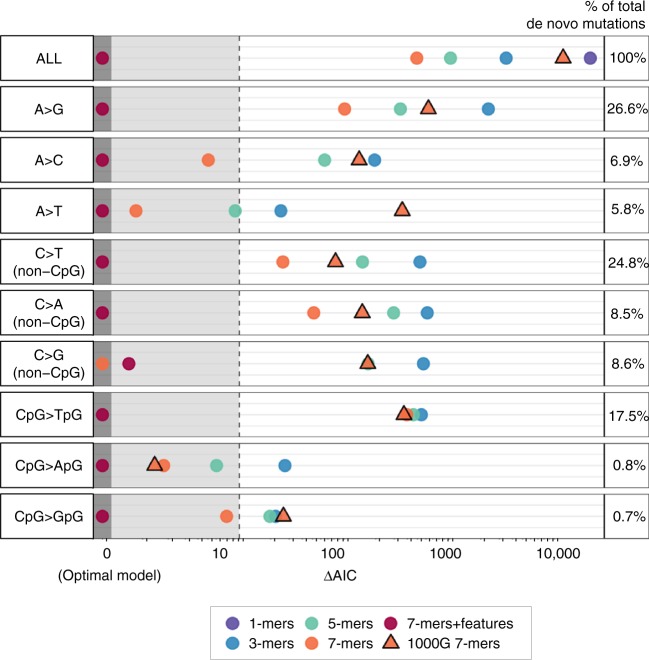
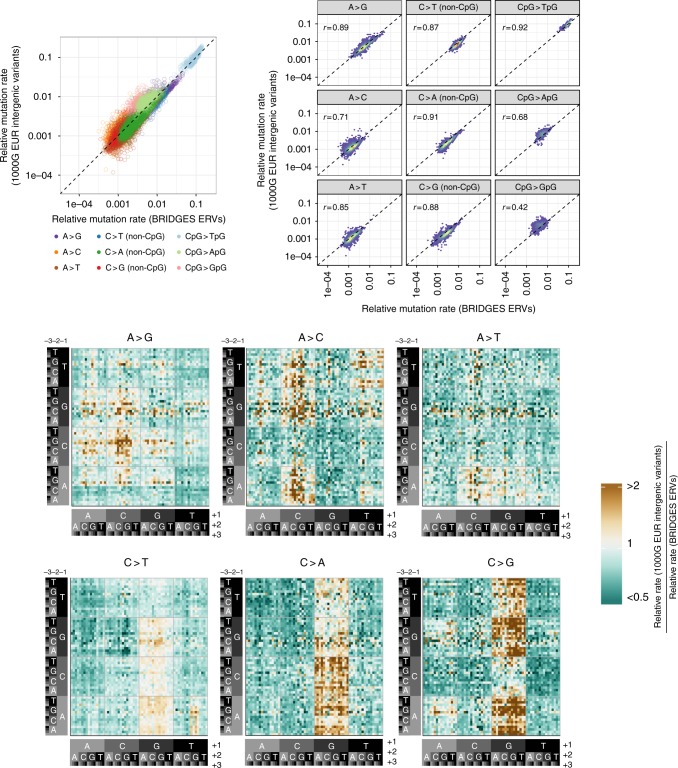
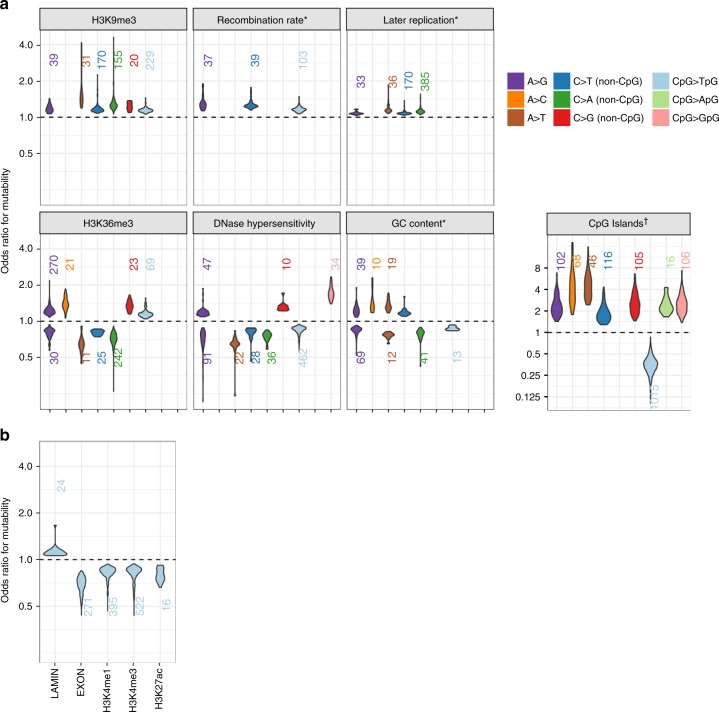
A detailed understanding of the genome-wide variability of single-nucleotide germline mutation rates is essential to studying human genome evolution. Here, we use ~36 million singleton variants from 3560 whole-genome sequences to infer fine-scale patterns of mutation rate heterogeneity. Mutability is jointly affected by adjacent nucleotide context and diverse genomic features of the surrounding region, including histone modifications, replication timing, and recombination rate, sometimes suggesting specific mutagenic mechanisms. Remarkably, GC content, DNase hypersensitivity, CpG islands, and H3K36 trimethylation are associated with both increased and decreased mutation rates depending on nucleotide context. We validate these estimated effects in an independent dataset of ~46,000 de novo mutations, and confirm our estimates are more accurate than previously published results based on ancestrally older variants without considering genomic features. Our results thus provide the most refined portrait to date of the factors contributing to genome-wide variability of the human germline mutation rate.
深入了解单核苷酸种系突变率的全基因组变异性对于研究人类基因组进化至关重要。在这里,我们使用来自 3560 个全基因组序列的约 3600 万个单倍体变体来推断突变率异质性的精细模式。易变性受到相邻核苷酸环境和周围区域的多种基因组特征的共同影响,包括组蛋白修饰、复制时间和重组率,有时表明存在特定的诱变机制。值得注意的是,GC 含量、DNase 超敏性、CpG 岛和 H3K36 三甲基化与核苷酸环境有关,既增加了突变率,也降低了突变率。我们在一个包含约 46000 个新突变的独立数据集对这些估计的效应进行了验证,并证实我们的估计比以前基于不考虑基因组特征的祖先更老的变体的发表结果更准确。因此,我们的结果提供了迄今为止最精细的人类种系突变率全基因组变异性的因素图谱。