Suzuki Shuhei, Okada Masashi, Takeda Hiroyuki, Kuramoto Kenta, Sanomachi Tomomi, Togashi Keita, Seino Shizuka, Yamamoto Masahiro, Yoshioka Takashi, Kitanaka Chifumi
Department of Molecular Cancer Science, Yamagata University School of Medicine, Yamagata 990-9585, Japan.
Department of Clinical Oncology, Yamagata University School of Medicine, Yamagata 990-9585, Japan.
Oncotarget. 2018 Aug 24;9(66):32667-32679. doi: 10.18632/oncotarget.25994.
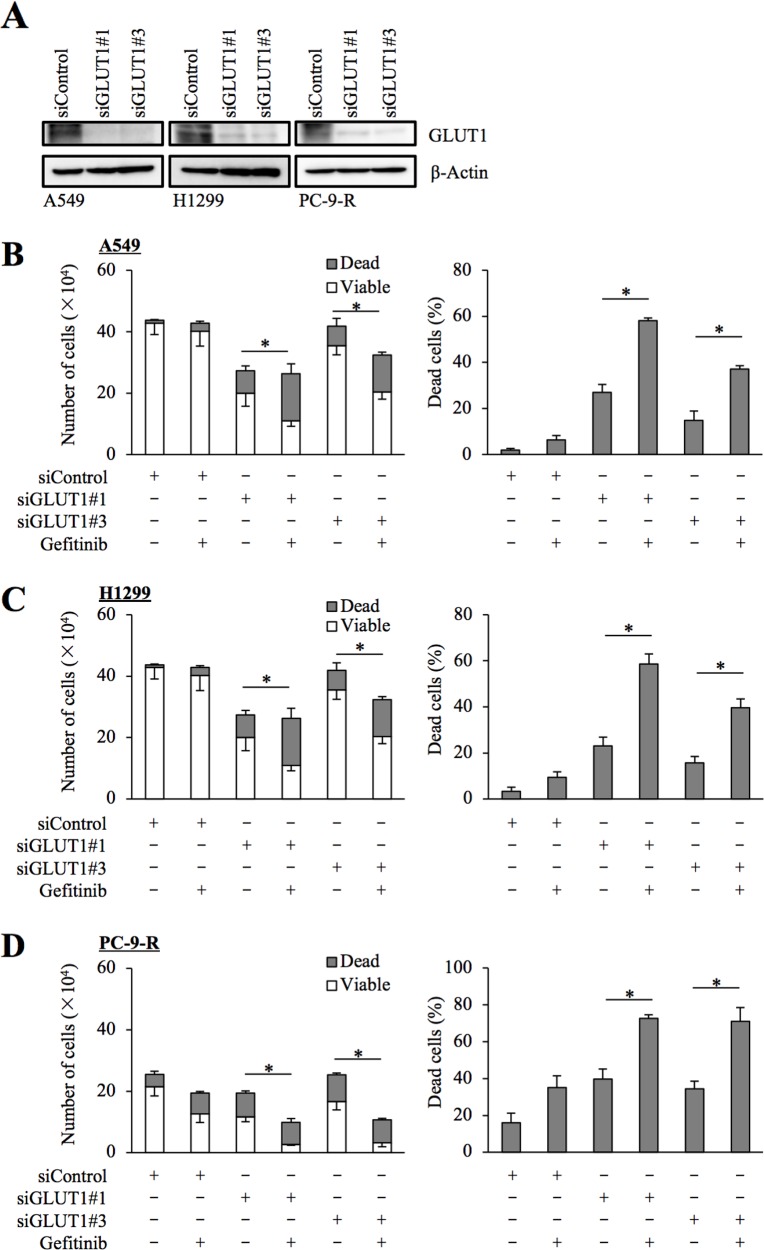
Use of epidermal growth factor receptor (EGFR) inhibitors represented by gefitinib and erlotinib has become the standard of treatment for non-small-cell lung cancers (NSCLCs) with activating EGFR mutations. However, the majority of NSCLCs, which overexpress EGFR without such mutations, are resistant to EGFR inhibitors, and the mechanism(s) behind such primary resistance of NSCLCs without activating EGFR mutations to EGFR inhibitors still remains poorly understood. Here in this study, we show that glucose metabolism mediated by GLUT1, a facilitative glucose transporter, is involved in gefitinib resistance of NSCLC cells. We found that GLUT1 expression and glucose uptake were increased in resistant NSCLC cells after gefitinib treatment and that genetic as well as pharmacological inhibition of GLUT1 sensitized not only NSCLC cells with primary resistance but also those with acquired resistance to gefitinib. , the combination of systemic gefitinib and a GLUT1 inhibitor, both of which failed to inhibit tumor growth when administered alone, significantly inhibited the growth of xenograft tumors formed by the implantation of NSCLC cells with wild-type EGFR (wt-EGFR). Since our data indicated that GLUT1 was similarly involved in erlotinib resistance, our findings suggest that the activity of GLUT1-mediated glucose metabolism could be a critical determinant for the sensitivity of NSCLC cells to EGFR inhibitors and that concurrent GLUT1 inhibition may therefore be a mechanism-based approach to treating NSCLCs resistant to EGFR inhibitors, including those with wt-EGFR.
以吉非替尼和厄洛替尼为代表的表皮生长因子受体(EGFR)抑制剂已成为治疗具有激活型EGFR突变的非小细胞肺癌(NSCLC)的标准疗法。然而,大多数过表达EGFR但无此类突变的NSCLC对EGFR抑制剂耐药,而无激活型EGFR突变的NSCLC对EGFR抑制剂产生这种原发性耐药的机制仍知之甚少。在本研究中,我们发现,由易化性葡萄糖转运蛋白GLUT1介导的糖代谢参与了NSCLC细胞对吉非替尼的耐药。我们发现,吉非替尼处理后,耐药NSCLC细胞中GLUT1表达和葡萄糖摄取增加,并且对GLUT1进行基因抑制以及药理学抑制不仅使原发性耐药的NSCLC细胞,也使获得性耐药的NSCLC细胞对吉非替尼敏感。此外,全身性吉非替尼与GLUT1抑制剂联合使用时,二者单独给药均无法抑制肿瘤生长,但却能显著抑制植入野生型EGFR(wt-EGFR)的NSCLC细胞形成的异种移植瘤的生长。由于我们的数据表明GLUT1同样参与了对厄洛替尼的耐药,我们的研究结果提示,GLUT1介导的糖代谢活性可能是NSCLC细胞对EGFR抑制剂敏感性的关键决定因素,因此,同时抑制GLUT1可能是一种基于机制的治疗对EGFR抑制剂耐药的NSCLC的方法,包括那些具有wt-EGFR的NSCLC。