Iijima Hiroyuki, Iwano Reiko, Tanaka Yukichi, Muroya Koji, Fukuda Tokiko, Sugie Hideo, Kurosawa Kenji, Adachi Masanori
Department of Endocrinology and Metabolism, Kanagawa Children's Medical Center, Mutsukawa 2-138-4, Minami-ku, Yokohama 232-8555, Japan.
Department of Pathology, Kanagawa Children's Medical Center, Mutsukawa 2-138-4, Minami-ku, Yokohama 232-8555, Japan.
Mol Genet Metab Rep. 2018 Sep 13;17:31-37. doi: 10.1016/j.ymgmr.2018.09.001. eCollection 2018 Dec.
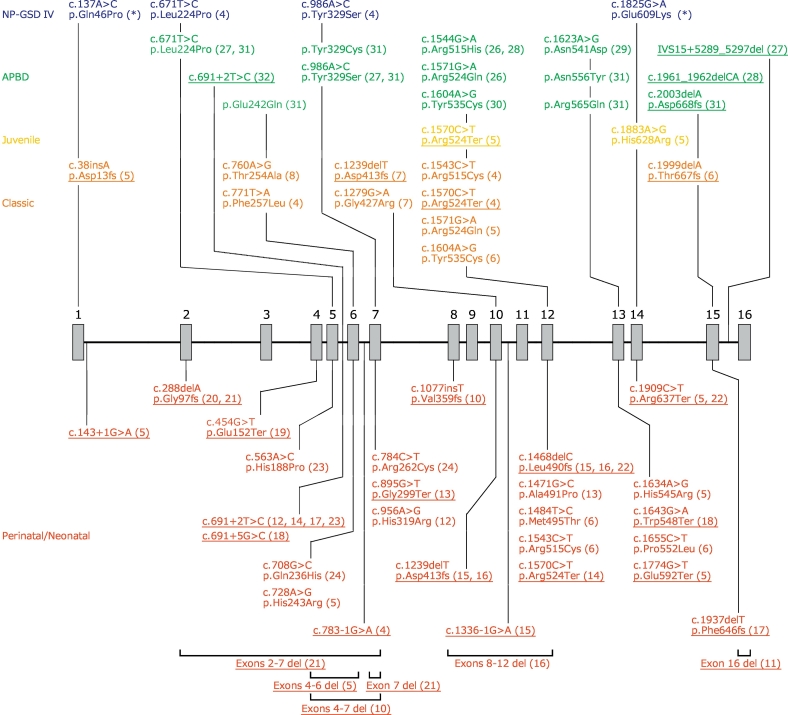
Glycogen storage disease type IV (GSD IV), caused by mutations, has a quite wide phenotypic variation. While the classic hepatic form and the perinatal/neonatal neuromuscular forms result in early mortality, milder manifestations include non-progressive form (NP-GSD IV) and adult polyglucosan body disease (APBD). Thus far, only one clinical case of a patient with compound heterozygous mutations has been reported for the molecular analysis of NP-GSD IV. This study aimed to elucidate the molecular basis in a NP-GSD IV patient via protein expression analysis and to obtain a clearer genotype-phenotype relationship in GSD IV.
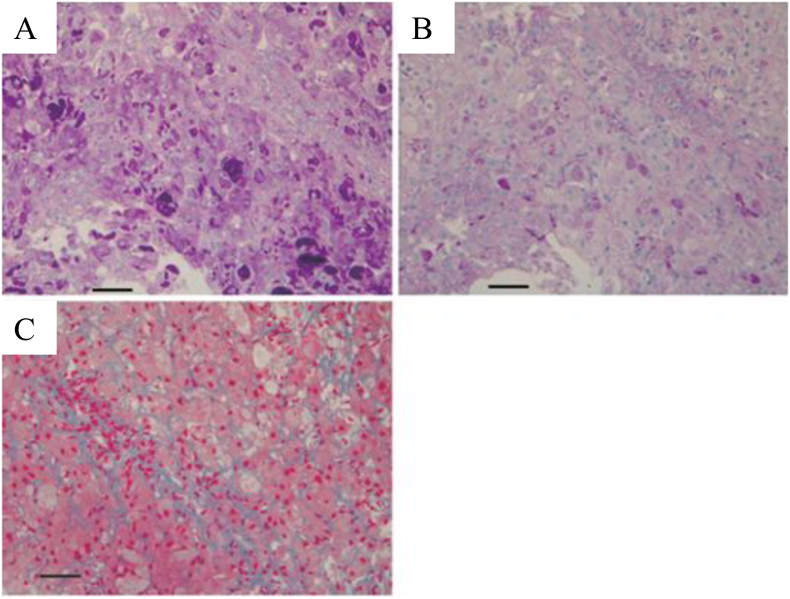
A Japanese boy presented hepatosplenomegaly at 2 years of age. Developmental delay, neurological symptoms, and cardiac dysfunction were not apparent. Observation of hepatocytes with periodic acid-Schiff-positive materials resistant to diastase, coupled with resolution of hepatosplenomegaly at 8 years of age, yielded a diagnosis of NP-GSD IV. Glycogen branching enzyme activity was decreased in erythrocytes. At 13 years of age, he developed epilepsy, which was successfully controlled by carbamazepine.
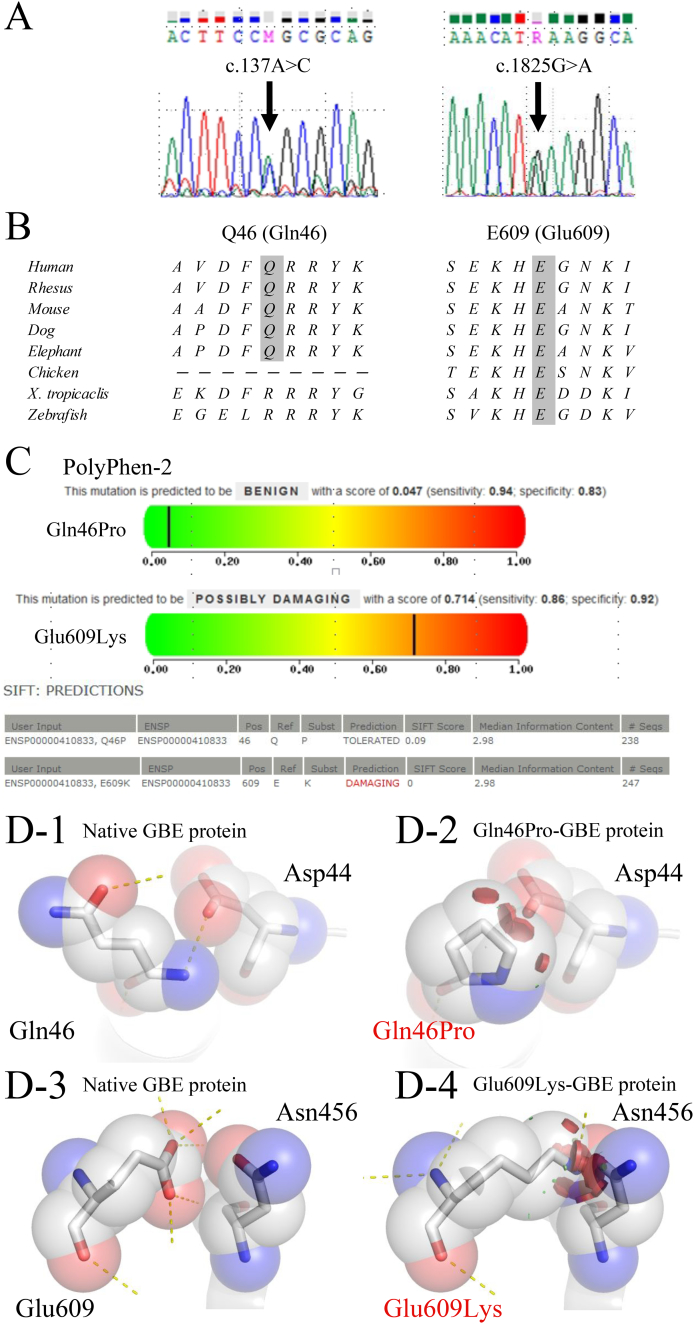
In this study, we identified compound heterozygous mutations (p.Gln46Pro and p.Glu609Lys). The branching activities of the mutant proteins expressed using were examined in a reaction with starch. The result showed that both mutants had approximately 50% activity of the wild type protein.
This is the second clinical report of a NP-GSD IV patient with a definite molecular elucidation. Based on the clinical and genotypic overlapping between NP-GSD IV and APBD, we suggest both are in a continuum.
糖原贮积病IV型(GSD IV)由基因突变引起,具有相当广泛的表型变异。虽然经典的肝型和围产期/新生儿神经肌肉型会导致早期死亡,但较轻的表现包括非进行性形式(NP-GSD IV)和成人多糖体病(APBD)。迄今为止,仅报道了一例复合杂合突变患者的临床病例用于NP-GSD IV的分子分析。本研究旨在通过蛋白质表达分析阐明一名NP-GSD IV患者的分子基础,并在GSD IV中获得更清晰的基因型-表型关系。
一名日本男孩在2岁时出现肝脾肿大。发育迟缓、神经症状和心脏功能障碍均不明显。观察到肝细胞内有对淀粉酶抵抗的过碘酸希夫阳性物质,且8岁时肝脾肿大消退,诊断为NP-GSD IV。红细胞中的糖原分支酶活性降低。13岁时,他患上癫痫,通过卡马西平成功控制。
在本研究中,我们鉴定出复合杂合突变(p.Gln46Pro和p.Glu609Lys)。使用[具体表达系统]表达的突变蛋白在与淀粉的反应中检测分支活性。结果显示,两种突变体的活性均约为野生型蛋白的50%。
这是第二例对NP-GSD IV患者进行明确分子阐释的临床报告。基于NP-GSD IV和APBD之间的临床和基因型重叠,我们认为两者处于连续统一体中。