Department of Endocrinology and Metabolism, Tianjin Medical University General Hospital, Tianjin, China.
Division of Metabolism, Endocrinology & Diabetes, University of Michigan Medical School, Ann Arbor, Michigan.
Diabetes Obes Metab. 2018 Sep;20 Suppl 2(Suppl 2):28-50. doi: 10.1111/dom.13378.
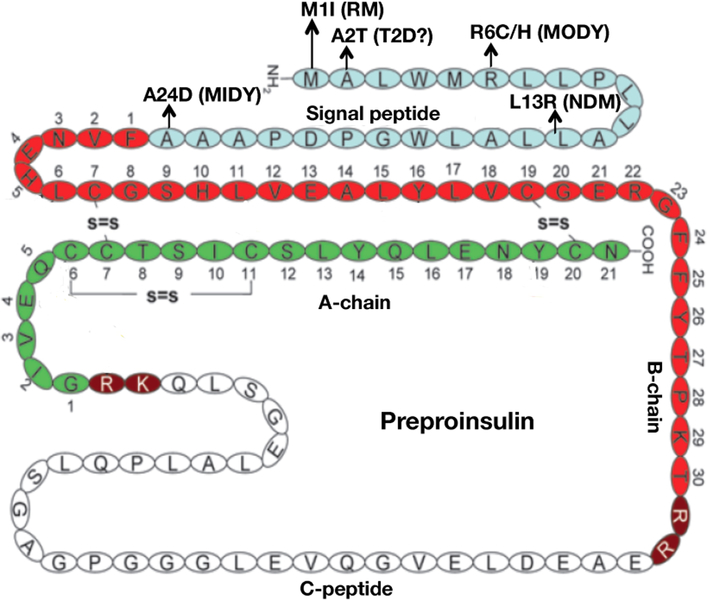
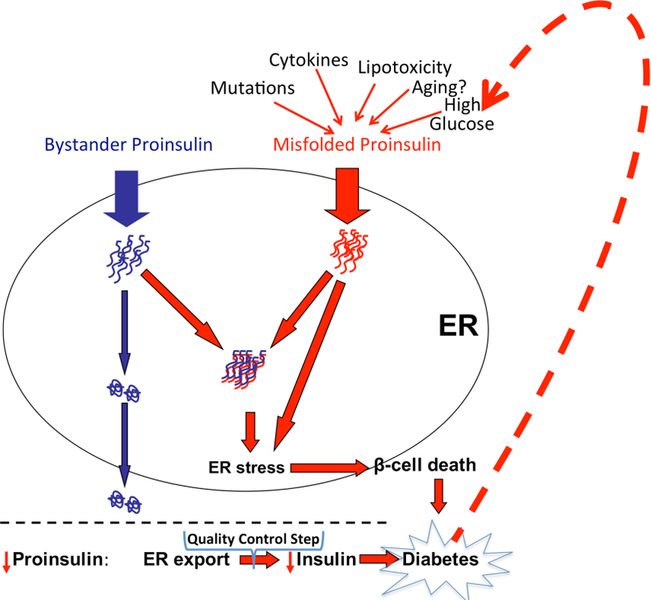
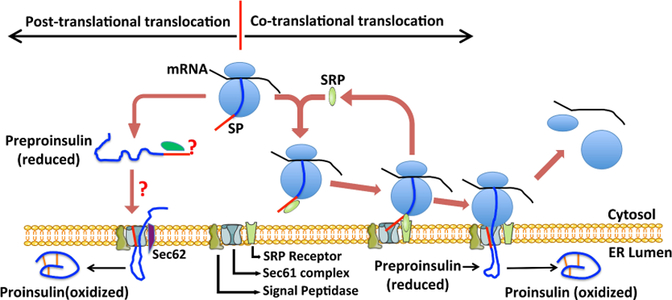
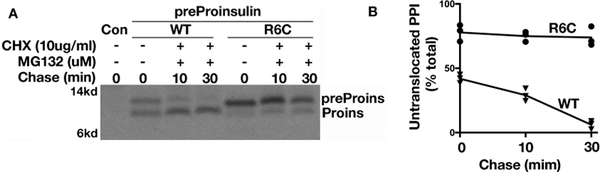
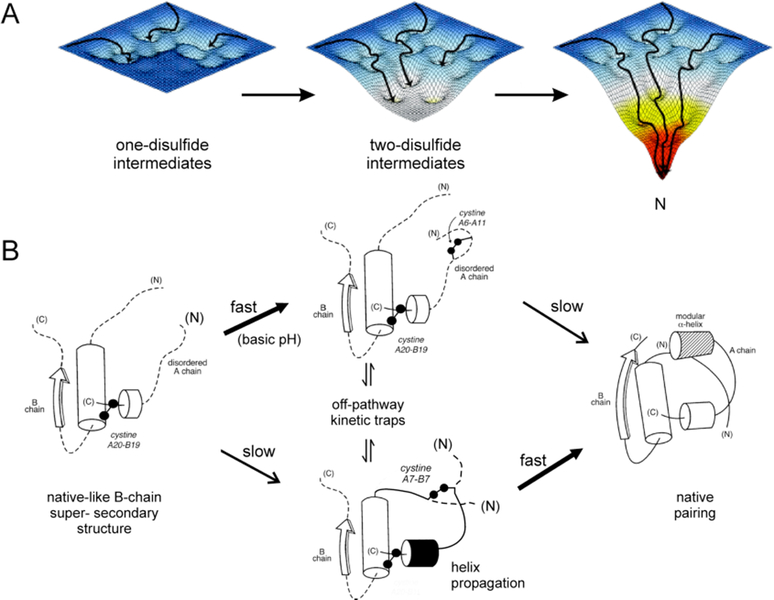
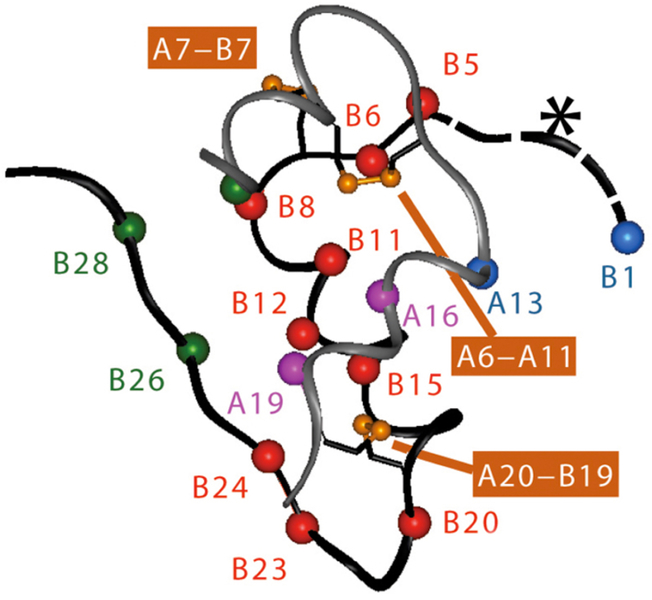
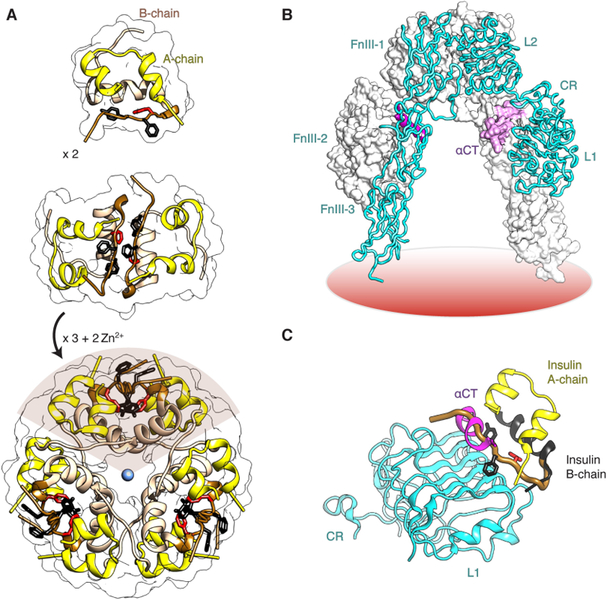
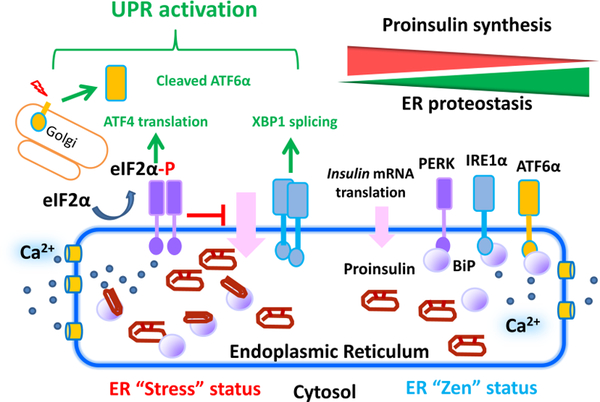
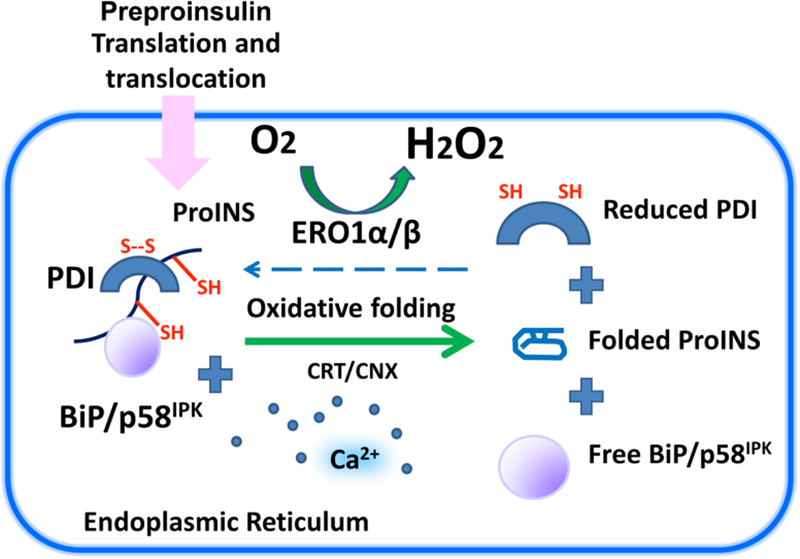
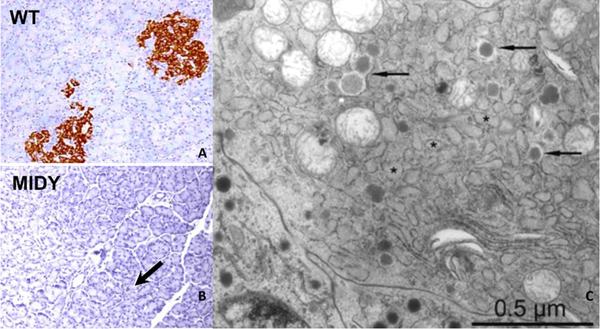
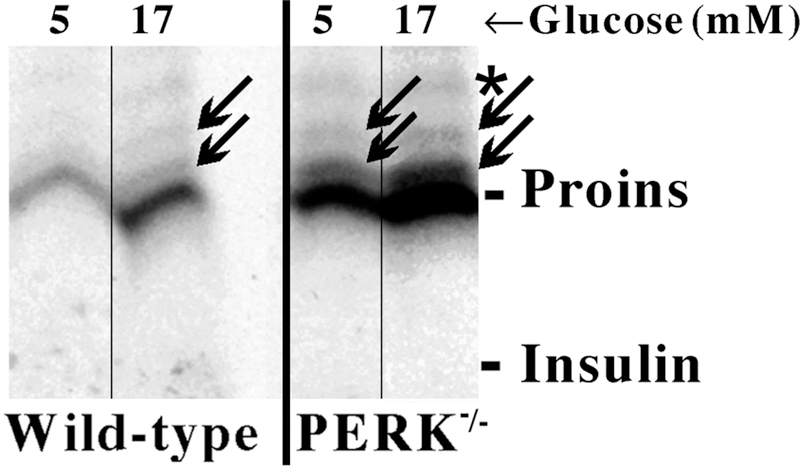
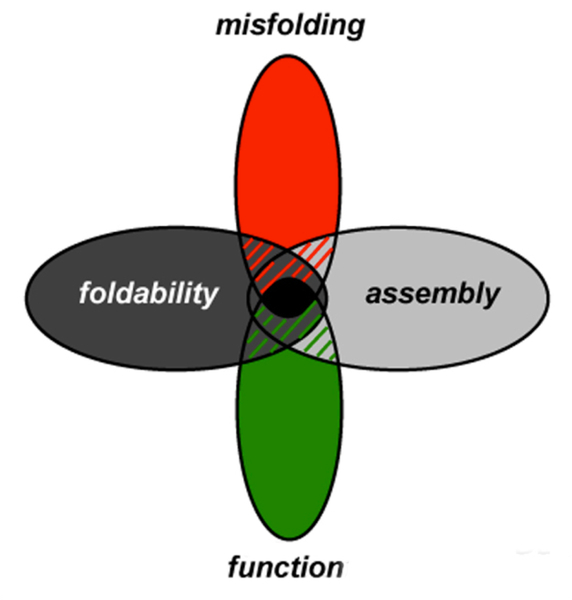
Insulin synthesis in pancreatic β-cells is initiated as preproinsulin. Prevailing glucose concentrations, which oscillate pre- and postprandially, exert major dynamic variation in preproinsulin biosynthesis. Accompanying upregulated translation of the insulin precursor includes elements of the endoplasmic reticulum (ER) translocation apparatus linked to successful orientation of the signal peptide, translocation and signal peptide cleavage of preproinsulin-all of which are necessary to initiate the pathway of proper proinsulin folding. Evolutionary pressures on the primary structure of proinsulin itself have preserved the efficiency of folding ("foldability"), and remarkably, these evolutionary pressures are distinct from those protecting the ultimate biological activity of insulin. Proinsulin foldability is manifest in the ER, in which the local environment is designed to assist in the overall load of proinsulin folding and to favour its disulphide bond formation (while limiting misfolding), all of which is closely tuned to ER stress response pathways that have complex (beneficial, as well as potentially damaging) effects on pancreatic β-cells. Proinsulin misfolding may occur as a consequence of exuberant proinsulin biosynthetic load in the ER, proinsulin coding sequence mutations, or genetic predispositions that lead to an altered ER folding environment. Proinsulin misfolding is a phenotype that is very much linked to deficient insulin production and diabetes, as is seen in a variety of contexts: rodent models bearing proinsulin-misfolding mutants, human patients with Mutant INS-gene-induced Diabetes of Youth (MIDY), animal models and human patients bearing mutations in critical ER resident proteins, and, quite possibly, in more common variety type 2 diabetes.
胰岛β细胞中的胰岛素合成起始于前胰岛素原。血糖浓度在餐前和餐后波动,对前胰岛素原生物合成产生主要的动态变化。伴随着胰岛素前体翻译的上调,包括与信号肽成功定向、前胰岛素原易位和信号肽切割相关的内质网(ER)易位装置的元件,所有这些都是启动正确胰岛素原折叠途径所必需的。前胰岛素原本身的一级结构所承受的进化压力保留了折叠的效率(“折叠性”),值得注意的是,这些进化压力与保护胰岛素最终生物活性的压力不同。前胰岛素原的折叠性在 ER 中表现出来,其中局部环境旨在协助前胰岛素原折叠的整体负荷,并有利于其二硫键形成(同时限制错误折叠),所有这些都与 ER 应激反应途径密切相关,这些途径对胰岛β细胞具有复杂的(有益的,以及潜在的有害的)影响。前胰岛素原错误折叠可能是由于 ER 中前胰岛素原生物合成负荷过多、前胰岛素原编码序列突变或遗传易感性导致 ER 折叠环境改变而发生的。前胰岛素原错误折叠是一种与胰岛素生成不足和糖尿病密切相关的表型,在各种情况下都有表现:携带前胰岛素原错误折叠突变体的啮齿动物模型、携带 Mutant INS-gene-induced Diabetes of Youth (MIDY) 的人类患者、携带关键 ER 驻留蛋白突变的动物模型和人类患者,以及在更常见的 2 型糖尿病中。