Department of Integrative Evolutionary Biology, Max-Planck-Institute for Developmental Biology, Max-Planck-Ring 9, 72076 Tübingen, Germany.
Genome Res. 2018 Nov;28(11):1664-1674. doi: 10.1101/gr.234971.118. Epub 2018 Sep 19.
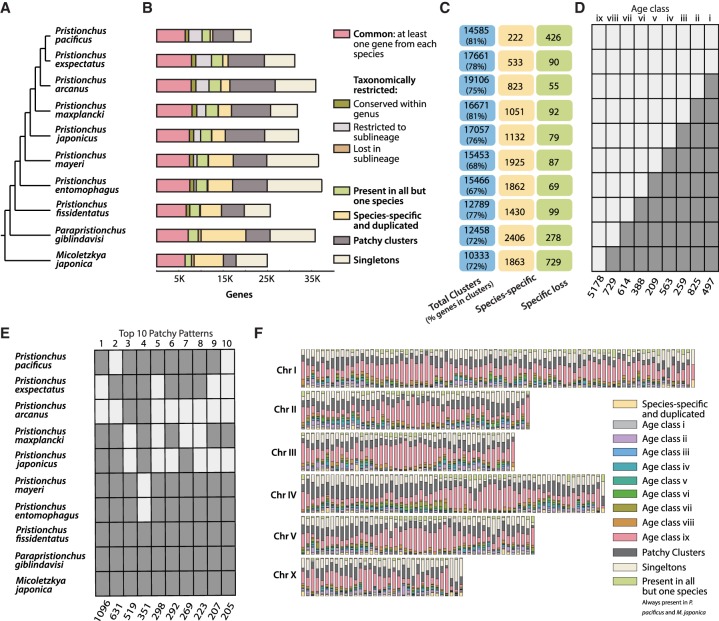
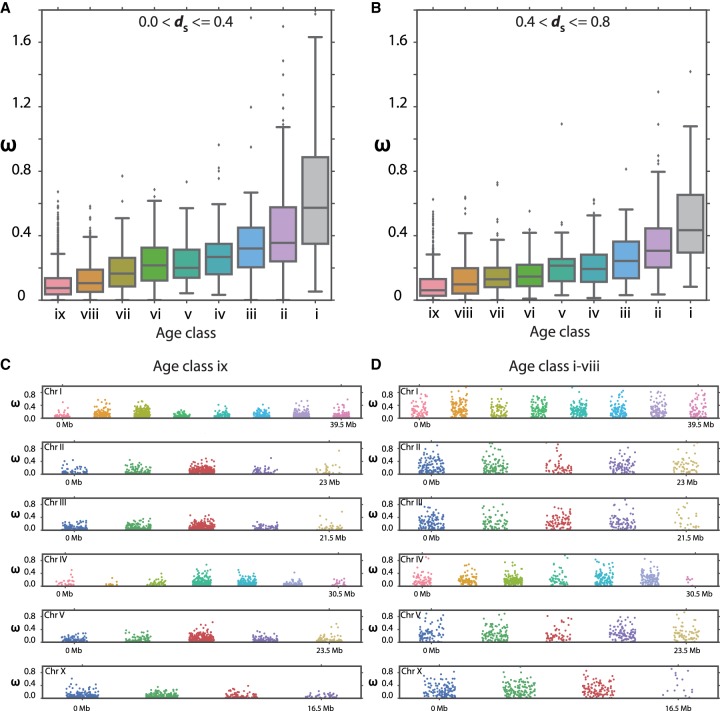
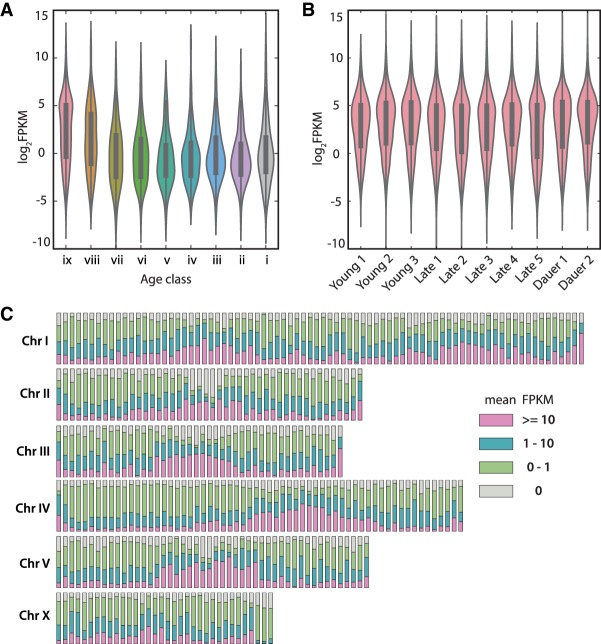
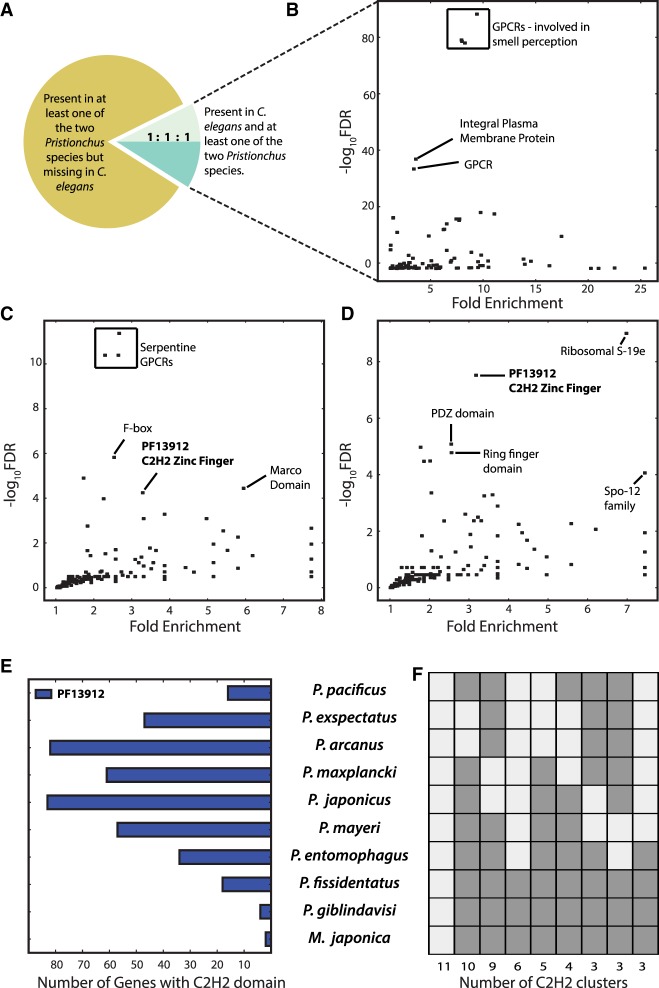
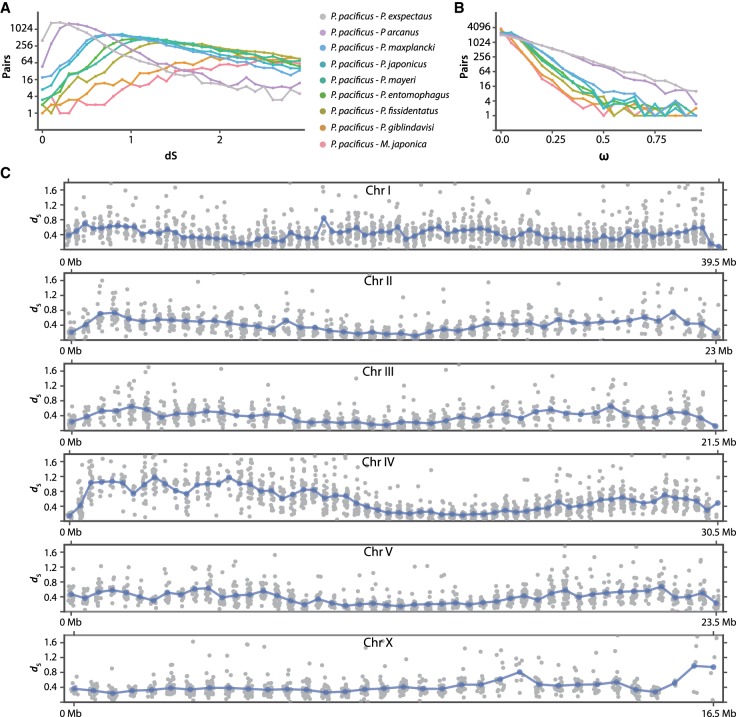
The widespread identification of genes without detectable homology in related taxa is a hallmark of genome sequencing projects in animals, together with the abundance of gene duplications. Such genes have been called novel, young, taxon-restricted, or orphans, but little is known about the mechanisms accounting for their origin, age, and mode of evolution. Phylogenomic studies relying on deep and systematic taxon sampling and using the comparative method can provide insight into the evolutionary dynamics acting on novel genes. We used a phylogenomic approach for the nematode model organism and sequenced six additional and two outgroup species. This resulted in 10 genomes with a ladder-like phylogeny, sequenced in one laboratory using the same platform and analyzed by the same bioinformatic procedures. Our analysis revealed that 68%-81% of genes are assignable to orthologous gene families, the majority of which defined nine age classes with presence/absence patterns that can be explained by single evolutionary events. Contrasting different age classes, we find that older age classes are concentrated at chromosome centers, whereas novel gene families preferentially arise at the periphery, are weakly expressed, evolve rapidly, and have a high propensity of being lost. Over time, they increase in expression and become more constrained. Thus, the detailed phylogenetic resolution allowed a comprehensive characterization of the evolutionary dynamics of genomes indicating that distribution of age classes and their associated differences shape chromosomal divergence. This study establishes the system for future research on the mechanisms that drive the formation of novel genes.
在动物的基因组测序项目中,广泛识别在相关分类单元中没有可检测到同源性的基因是一个标志,同时还存在大量的基因重复。这些基因被称为新的、年轻的、分类单元受限的或孤儿基因,但对于它们的起源、年龄和进化模式的机制知之甚少。依赖于深度和系统的分类单元采样并使用比较方法的系统基因组学研究可以深入了解作用于新基因的进化动态。我们使用线虫模式生物进行了系统基因组学研究,并对另外 6 个和 2 个外群物种进行了测序。这导致了 10 个具有阶梯状系统发育的基因组,在一个实验室中使用相同的平台进行测序,并通过相同的生物信息学程序进行分析。我们的分析表明,68%-81%的基因可分配到同源基因家族,其中大多数基因定义了九个年龄类,其存在/缺失模式可以用单个进化事件来解释。对比不同的年龄类,我们发现较老的年龄类集中在染色体中心,而新的基因家族则优先出现在染色体边缘,表达较弱,进化迅速,并且有很高的丢失倾向。随着时间的推移,它们的表达增加并且变得更受限制。因此,详细的系统发育分辨率允许对 基因组的进化动态进行全面描述,表明年龄类别的分布及其相关差异塑造了染色体的分化。这项研究为未来研究驱动新基因形成的机制奠定了基础。