Anslan Sten, Nilsson R Henrik, Wurzbacher Christian, Baldrian Petr, Bahram Mohammad
Braunschweig University of Technology, Zoological Institute, Mendelssohnstr. 4, 38106 Braunschweig, Germany.
Gothenburg Global Biodiversity Centre, Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Gothenburg, Sweden.
MycoKeys. 2018 Sep 11(39):29-40. doi: 10.3897/mycokeys.39.28109. eCollection 2018.
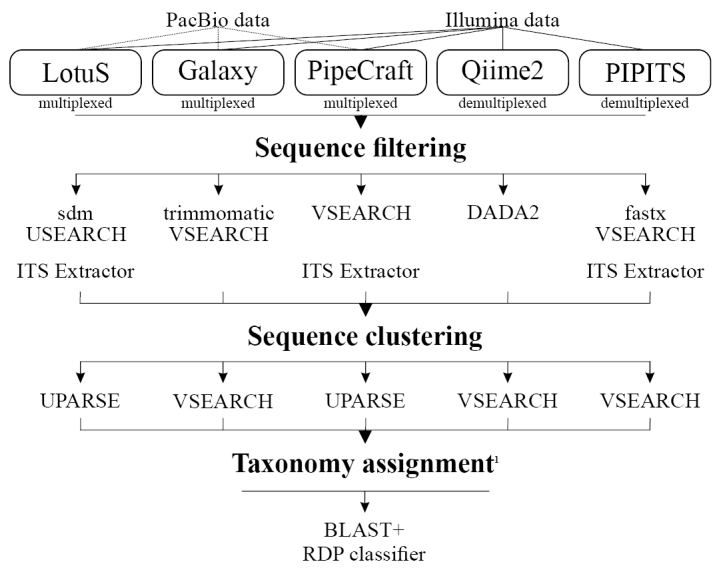
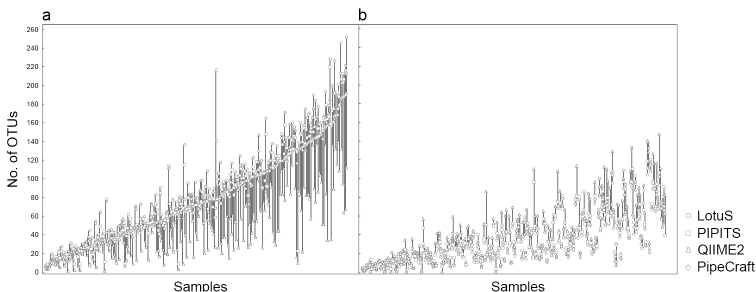
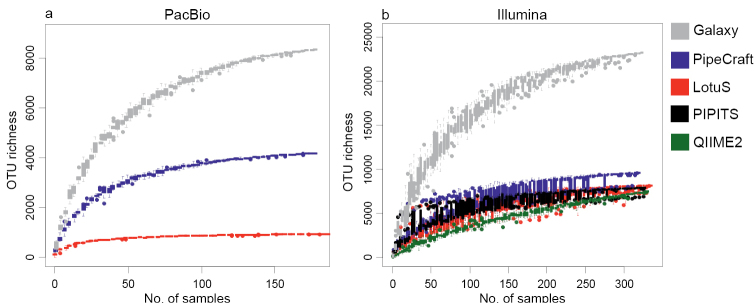
Along with recent developments in high-throughput sequencing (HTS) technologies and thus fast accumulation of HTS data, there has been a growing need and interest for developing tools for HTS data processing and communication. In particular, a number of bioinformatics tools have been designed for analysing metabarcoding data, each with specific features, assumptions and outputs. To evaluate the potential effect of the application of different bioinformatics workflow on the results, we compared the performance of different analysis platforms on two contrasting high-throughput sequencing data sets. Our analysis revealed that the computation time, quality of error filtering and hence output of specific bioinformatics process largely depends on the platform used. Our results show that none of the bioinformatics workflows appears to perfectly filter out the accumulated errors and generate Operational Taxonomic Units, although PipeCraft, LotuS and PIPITS perform better than QIIME2 and Galaxy for the tested fungal amplicon dataset. We conclude that the output of each platform requires manual validation of the OTUs by examining the taxonomy assignment values.
随着高通量测序(HTS)技术的最新发展以及HTS数据的快速积累,开发用于HTS数据处理和通信的工具的需求和兴趣日益增长。特别是,已经设计了许多生物信息学工具来分析元条形码数据,每个工具都有特定的特征、假设和输出。为了评估不同生物信息学工作流程的应用对结果的潜在影响,我们在两个对比鲜明的高通量测序数据集上比较了不同分析平台的性能。我们的分析表明,计算时间、错误过滤质量以及特定生物信息学过程的输出在很大程度上取决于所使用的平台。我们的结果表明,尽管对于测试的真菌扩增子数据集,PipeCraft、LotuS和PIPITS的表现优于QIIME2和Galaxy,但没有一个生物信息学工作流程似乎能完美地过滤掉累积的错误并生成操作分类单元。我们得出结论,每个平台的输出都需要通过检查分类学分配值来对操作分类单元进行人工验证。