Department of Medicine, Division of Gastroenterology and Hepatology, and Microbiome Core Facility, Center for Gastrointestinal Biology and Disease, School of Medicine, University of North Carolina, Campus Box 7555, 332 Isaac Taylor Hall, Chapel Hill, NC, 27599-7545, USA.
Laboratory of Biochemistry & Immunology, Faculty of Sciences, Mohammed V University, Rabat, Morocco.
BMC Microbiol. 2017 Sep 13;17(1):194. doi: 10.1186/s12866-017-1101-8.
Advancements in Next Generation Sequencing (NGS) technologies regarding throughput, read length and accuracy had a major impact on microbiome research by significantly improving 16S rRNA amplicon sequencing. As rapid improvements in sequencing platforms and new data analysis pipelines are introduced, it is essential to evaluate their capabilities in specific applications. The aim of this study was to assess whether the same project-specific biological conclusions regarding microbiome composition could be reached using different sequencing platforms and bioinformatics pipelines.
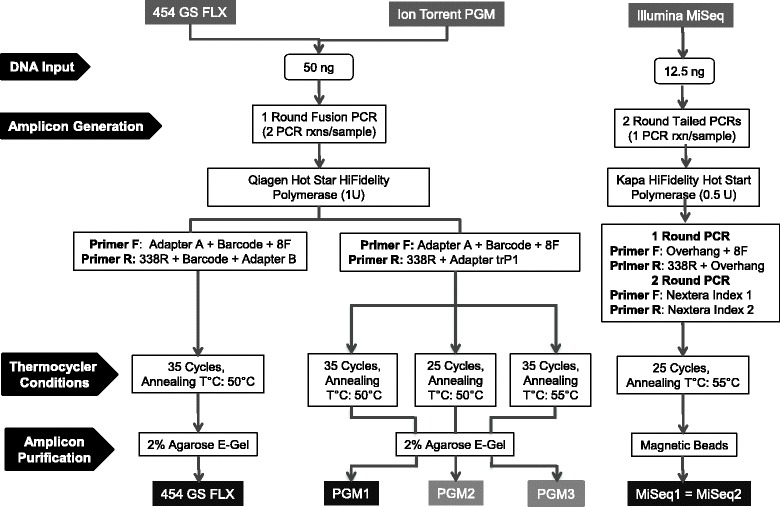
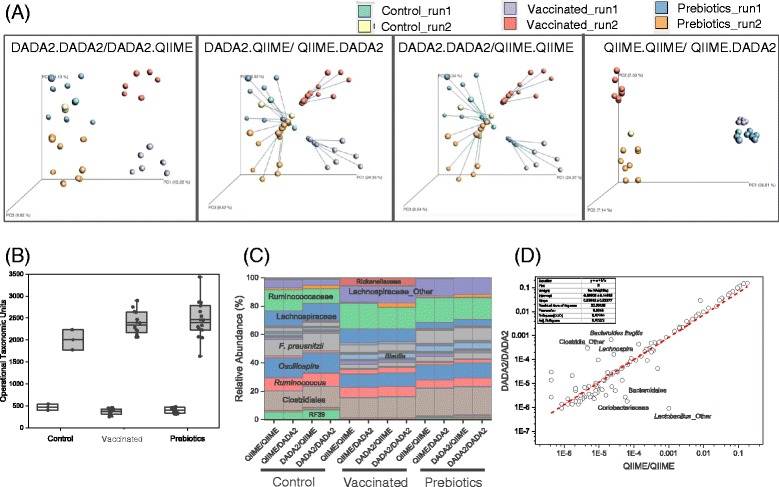
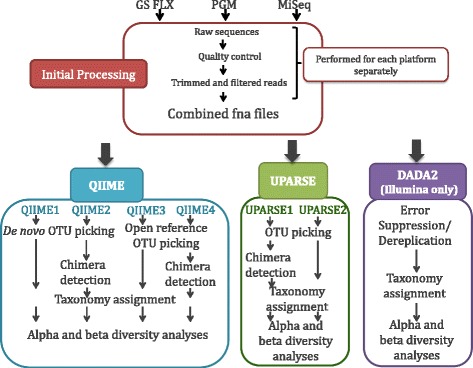
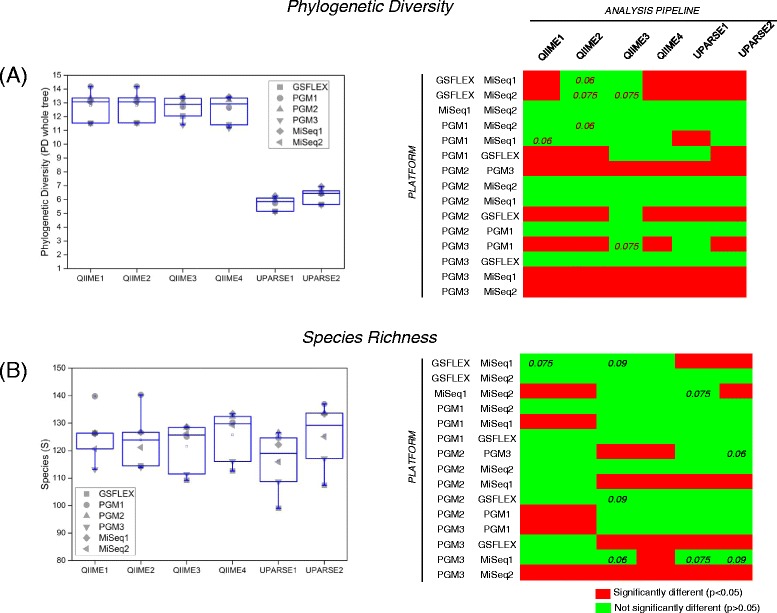
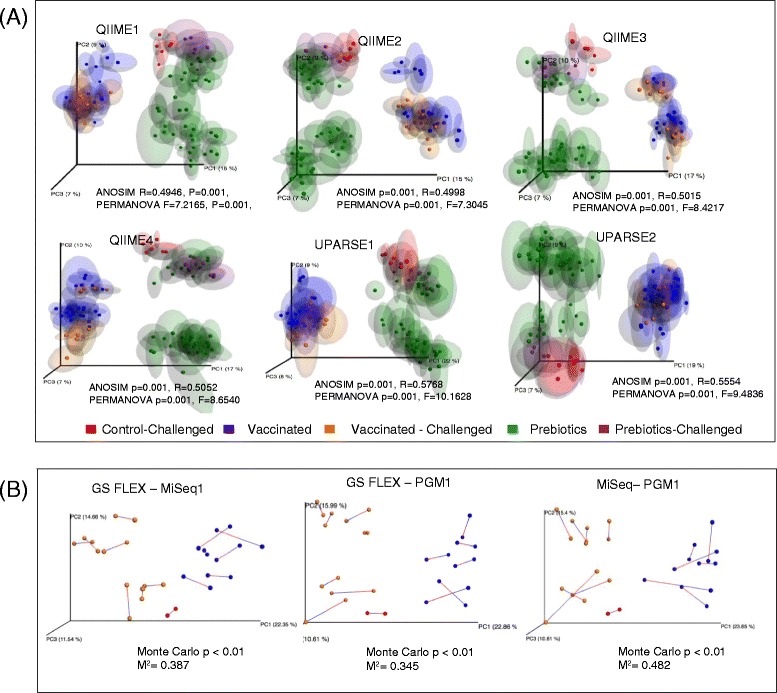
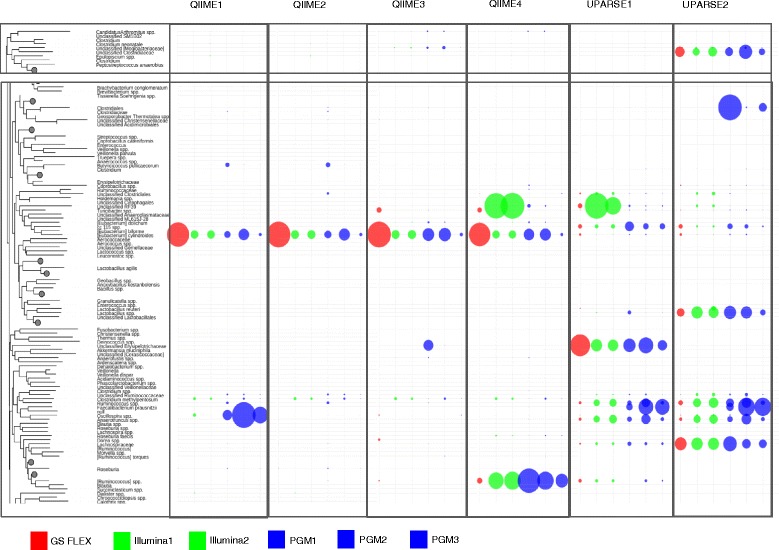
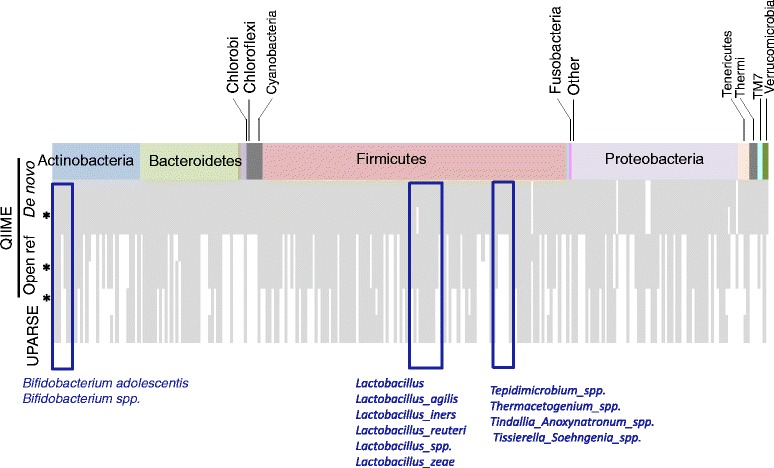
Chicken cecum microbiome was analyzed by 16S rRNA amplicon sequencing using Illumina MiSeq, Ion Torrent PGM, and Roche 454 GS FLX Titanium platforms, with standard and modified protocols for library preparation. We labeled the bioinformatics pipelines included in our analysis QIIME1 and QIIME2 (de novo OTU picking [not to be confused with QIIME version 2 commonly referred to as QIIME2]), QIIME3 and QIIME4 (open reference OTU picking), UPARSE1 and UPARSE2 (each pair differs only in the use of chimera depletion methods), and DADA2 (for Illumina data only). GS FLX+ yielded the longest reads and highest quality scores, while MiSeq generated the largest number of reads after quality filtering. Declines in quality scores were observed starting at bases 150-199 for GS FLX+ and bases 90-99 for MiSeq. Scores were stable for PGM-generated data. Overall microbiome compositional profiles were comparable between platforms; however, average relative abundance of specific taxa varied depending on sequencing platform, library preparation method, and bioinformatics analysis. Specifically, QIIME with de novo OTU picking yielded the highest number of unique species and alpha diversity was reduced with UPARSE and DADA2 compared to QIIME.
The three platforms compared in this study were capable of discriminating samples by treatment, despite differences in diversity and abundance, leading to similar biological conclusions. Our results demonstrate that while there were differences in depth of coverage and phylogenetic diversity, all workflows revealed comparable treatment effects on microbial diversity. To increase reproducibility and reliability and to retain consistency between similar studies, it is important to consider the impact on data quality and relative abundance of taxa when selecting NGS platforms and analysis tools for microbiome studies.
下一代测序(NGS)技术在通量、读长和准确性方面的进步,通过显著提高 16S rRNA 扩增子测序,对微生物组研究产生了重大影响。随着测序平台和新数据分析管道的快速改进,评估它们在特定应用中的能力至关重要。本研究旨在评估使用不同测序平台和生物信息学管道是否可以得出关于微生物组组成的相同特定项目生物学结论。
使用 Illumina MiSeq、Ion Torrent PGM 和 Roche 454 GS FLX Titanium 平台,通过标准和改良的文库制备方案,对鸡盲肠微生物组进行了 16S rRNA 扩增子测序。我们标记了包含在我们分析中的生物信息学管道 QIIME1 和 QIIME2(从头 OTU 挑选[与通常称为 QIIME2 的 QIIME 版本 2 不同,后者是指 QIIME2])、QIIME3 和 QIIME4(开放参考 OTU 挑选)、UPARSE1 和 UPARSE2(每对仅在使用嵌合体耗竭方法方面有所不同)以及 DADA2(仅用于 Illumina 数据)。GS FLX+ 产生的读长最长,质量得分最高,而 MiSeq 在质量过滤后产生的读长最多。对于 GS FLX+,从第 150-199 个碱基开始,对于 MiSeq,从第 90-99 个碱基开始,质量得分开始下降。PGM 生成的数据得分稳定。尽管平台之间的总体微生物组组成谱相似,但特定类群的平均相对丰度取决于测序平台、文库制备方法和生物信息学分析。具体而言,与 QIIME 相比,具有从头 OTU 挑选的 QIIME 产生的独特物种数量最多,而 UPARSE 和 DADA2 降低了 alpha 多样性。
尽管在多样性和丰度方面存在差异,但本研究中比较的三个平台能够通过处理区分样本,导致相似的生物学结论。我们的结果表明,虽然在覆盖深度和系统发育多样性方面存在差异,但所有工作流程都揭示了类似的处理对微生物多样性的影响。为了提高重现性和可靠性,并在类似的研究中保持一致性,在选择微生物组研究的 NGS 平台和分析工具时,考虑数据质量和分类群相对丰度的影响非常重要。