Center for Occupational and Environmental Health, Department of Medicine, University of California, Irvine, California.
Molecular and Cell Biology, University of California, Merced, California.
Glia. 2018 Dec;66(12):2700-2718. doi: 10.1002/glia.23522. Epub 2018 Sep 12.
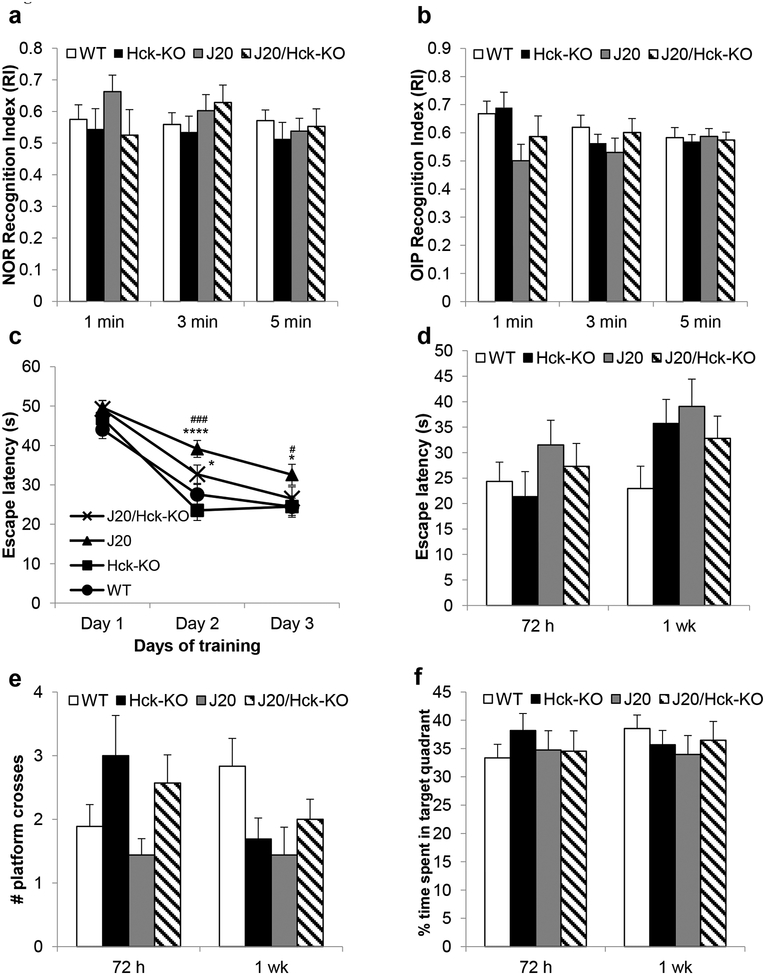
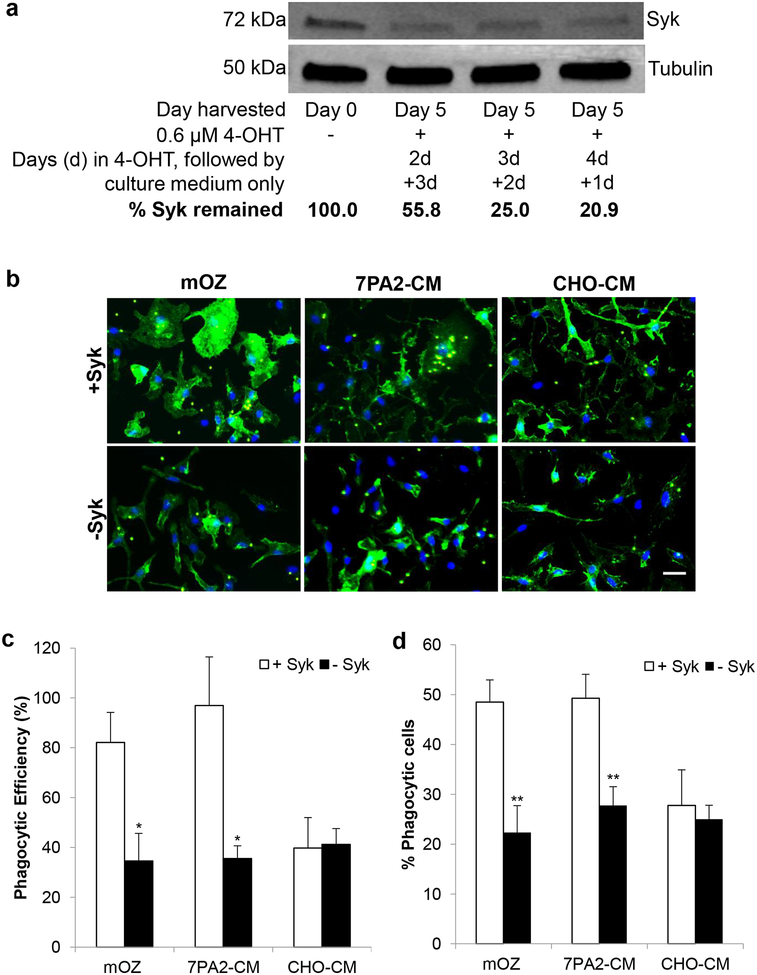
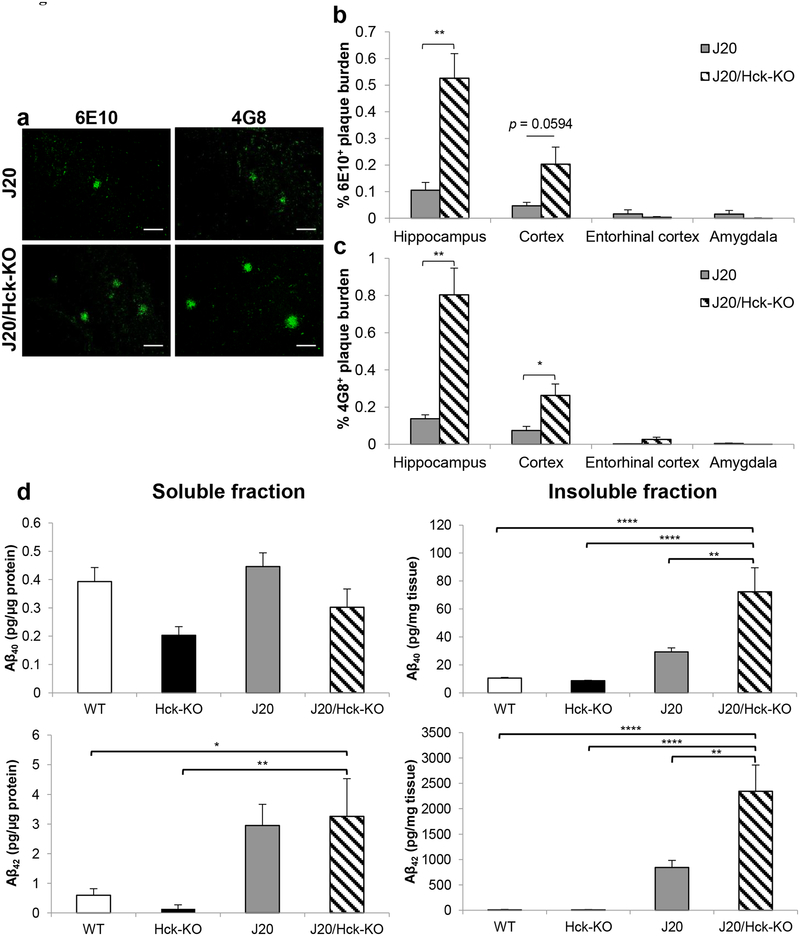
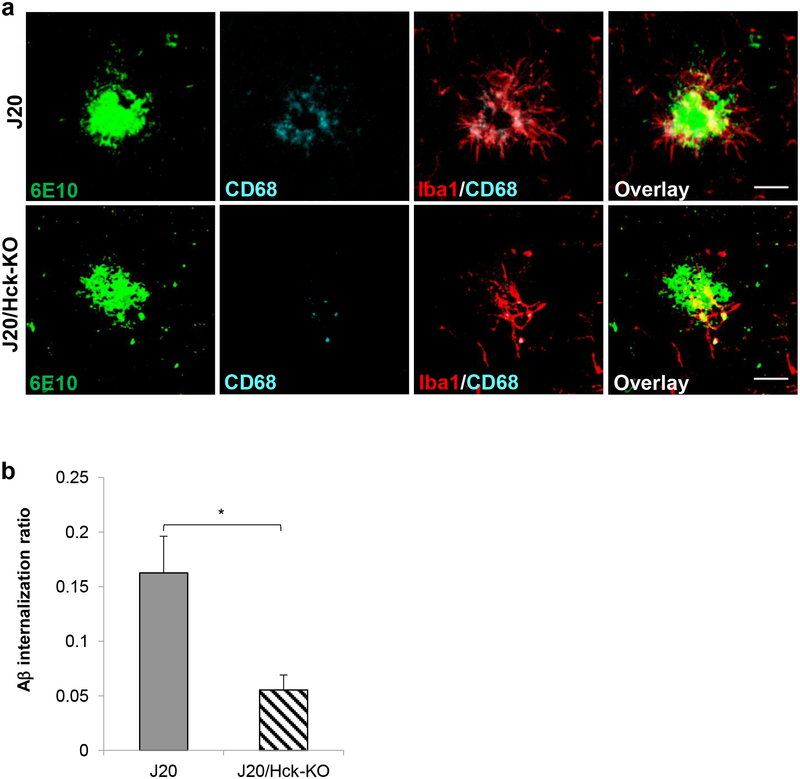
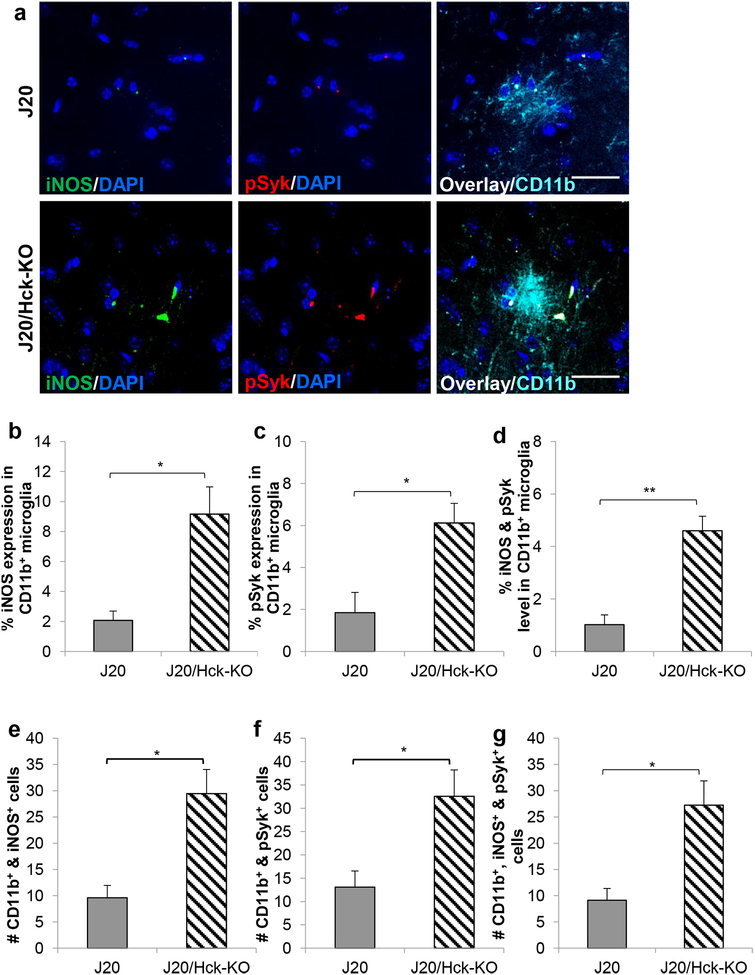
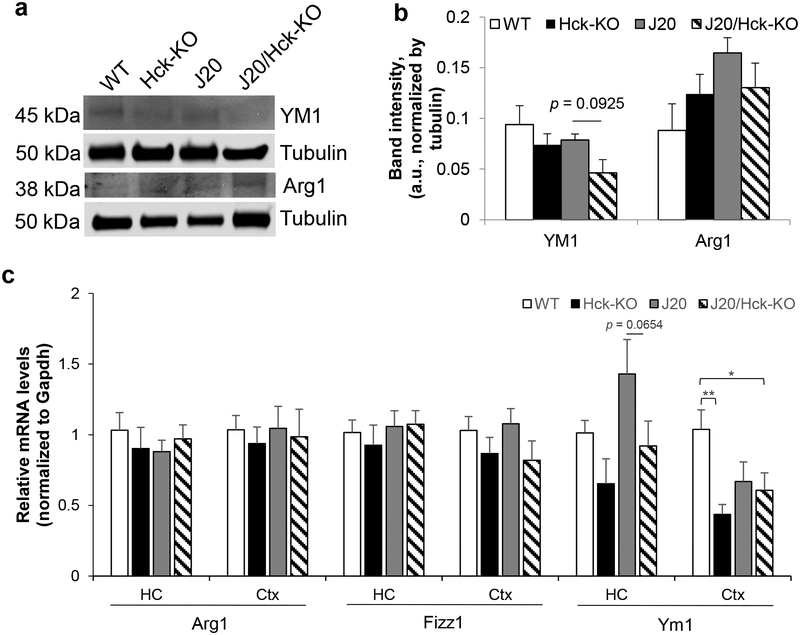
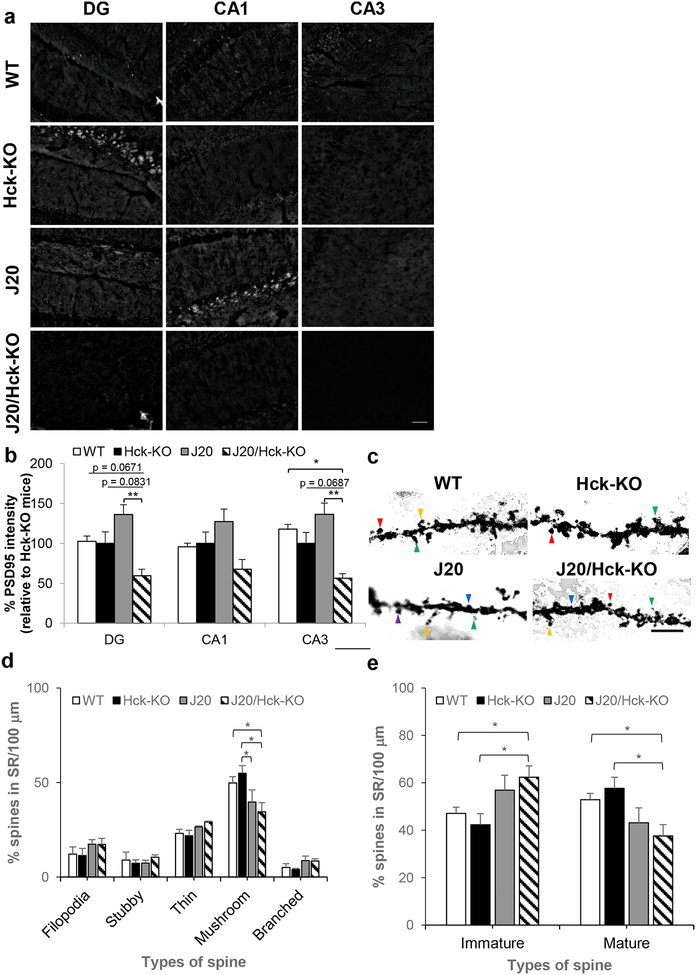
Emerging evidence have posited that dysregulated microglia impair clearance and containment of amyloid-β (Aβ) species in the brain, resulting in aberrant buildup of Aβ and onset of Alzheimer's disease (AD). Hematopoietic cell kinase (Hck) is one of the key regulators of phagocytosis among the Src family tyrosine kinases (SFKs) in myeloid cells, and its expression is found to be significantly altered in AD brains. However, the role of Hck signaling in AD pathogenesis is unknown. We employed pharmacological inhibition and genetic ablation of Hck in BV2 microglial cells and J20 mouse model of AD, respectively, to evaluate the impact of Hck deficiency on Aβ-stimulated microglial phagocytosis, Aβ clearance, and resultant AD-like neuropathology. Our in vitro data reveal that pharmacological inhibition of SFKs/Hck in BV2 cells and genetic ablation of their downstream kinase, spleen tyrosine kinase (Syk), in primary microglia significantly attenuate Aβ oligomers-stimulated microglial phagocytosis. Whereas in Hck-deficient J20 mice, we observed exacerbated Aβ plaque burden, reduced microglial coverage, containment, and phagocytosis of Aβ plaques, and induced iNOS expression in plaque-associated microglial clusters. These multifactorial changes in microglial activities led to attenuated PSD95 levels in hippocampal DG and CA3 regions, but did not alter the postsynaptic dendritic spine morphology at the CA1 region nor cognitive function of the mice. Hck inhibition thus accelerates early stage AD-like neuropathology by dysregulating microglial function and inducing neuroinflammation. Our data implicate that Hck pathway plays a prominent role in regulating microglial neuroprotective function during the early stage of AD development.
新出现的证据表明,失调的小胶质细胞会损害大脑中淀粉样β(Aβ)的清除和控制,导致 Aβ的异常积累,并引发阿尔茨海默病(AD)。造血细胞激酶(Hck)是髓样细胞中 Src 家族酪氨酸激酶(SFKs)中吞噬作用的关键调节因子之一,其在 AD 脑中的表达被发现显著改变。然而,Hck 信号通路在 AD 发病机制中的作用尚不清楚。我们分别在 BV2 小胶质细胞和 J20 AD 小鼠模型中使用 Hck 的药理学抑制和基因敲除来评估 Hck 缺乏对 Aβ刺激的小胶质细胞吞噬作用、Aβ清除以及由此产生的 AD 样神经病理学的影响。我们的体外数据显示,BV2 细胞中 SFKs/Hck 的药理学抑制和原代小胶质细胞中其下游激酶脾酪氨酸激酶(Syk)的基因敲除显著减弱了 Aβ 寡聚体刺激的小胶质细胞吞噬作用。而在 Hck 缺陷的 J20 小鼠中,我们观察到 Aβ 斑块负担增加,小胶质细胞覆盖、控制和 Aβ 斑块吞噬作用减少,以及斑块相关小胶质细胞簇中诱导型一氧化氮合酶(iNOS)的表达增加。这些小胶质细胞活性的多种变化导致海马齿状回和 CA3 区 PSD95 水平降低,但不改变 CA1 区的突触后树突棘形态或小鼠的认知功能。Hck 抑制通过调节小胶质细胞功能和诱导神经炎症,从而加速早期 AD 样神经病理学。我们的数据表明,Hck 通路在调节 AD 早期发展中小胶质细胞神经保护功能方面发挥着重要作用。