Department of Medical Biosciences, Umeå University, 90186, Umeå, Sweden.
Research Laboratory for Stereology and Neuroscience, Department of Neurology, Faculty of Health, Bispebjerg-Frederiksberg Hospital Copenhagen, and Institute of Clinical Medicine, University of Copenhagen, Copenhagen, Denmark.
Acta Neuropathol. 2018 Dec;136(6):939-953. doi: 10.1007/s00401-018-1915-y. Epub 2018 Oct 3.
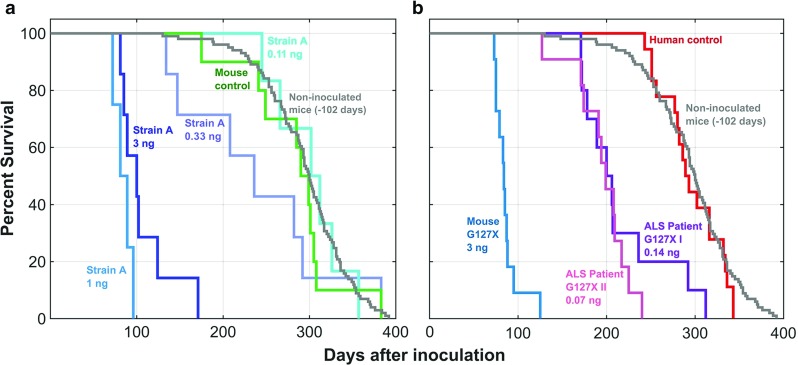
Motor neurons containing aggregates of superoxide dismutase 1 (SOD1) are hallmarks of amyotrophic lateral sclerosis (ALS) caused by mutations in the gene encoding SOD1. We have previously reported that two strains of mutant human (h) SOD1 aggregates (denoted A and B) can arise in hSOD1-transgenic models for ALS and that inoculation of such aggregates into the lumbar spinal cord of mice results in rostrally spreading, templated hSOD1 aggregation and premature fatal ALS-like disease. Here, we explored whether mutant hSOD1 aggregates with prion-like properties also exist in human ALS. Aggregate seeds were prepared from spinal cords from an ALS patient carrying the hSOD1 truncation mutation and from mice transgenic for the same mutation. To separate from mono-, di- or any oligomeric hSOD1 species, the seed preparation protocol included ultracentrifugation through a density cushion. The core structure of hSOD1 aggregates present in mice was strain A-like. Inoculation of the patient- or mouse-derived seeds into lumbar spinal cord of adult hSOD1-expressing mice induced strain A aggregation propagating along the neuraxis and premature fatal ALS-like disease (p < 0.0001). Inoculation of human or murine control seeds had no effect. The potencies of the ALS patient-derived seed preparations were high and disease was initiated in the transgenic mice by levels of hSOD1 aggregates much lower than those found in the motor system of patients carrying the mutation. The results suggest that prion-like growth and spread of hSOD1 aggregation could be the primary pathogenic mechanism, not only in hSOD1 transgenic rodent models, but also in human ALS.
含有超氧化物歧化酶 1 (SOD1) 聚集物的运动神经元是由 SOD1 基因编码突变引起的肌萎缩侧索硬化症 (ALS) 的标志。我们之前曾报道过,两种突变型人(h)SOD1 聚集体(分别表示为 A 和 B)可在 ALS 的 hSOD1 转基因模型中出现,并且将此类聚集体接种到小鼠的腰椎脊髓中会导致 rostrally 扩散、模板化的 hSOD1 聚集和过早致命的 ALS 样疾病。在这里,我们探讨了是否具有类朊病毒特性的突变型 hSOD1 聚集体也存在于人类 ALS 中。聚集体种子是从携带 hSOD1 截断突变的 ALS 患者的脊髓和携带相同突变的小鼠转基因中制备的。为了与单、二聚体或任何寡聚体 hSOD1 分离,种子制备方案包括通过密度垫超速离心。存在于小鼠中的 hSOD1 聚集体的核心结构类似于 A 型。将源自患者或小鼠的种子接种到表达 hSOD1 的成年小鼠的腰椎脊髓中,可诱导 A 型聚集物沿神经元轴突传播,并引发过早致命的 ALS 样疾病(p <0.0001)。接种人类或鼠对照种子没有影响。来自 ALS 患者的种子制剂的效力很高,并且通过低于携带突变的患者运动系统中发现的水平的 hSOD1 聚集体,在转基因小鼠中引发了疾病。这些结果表明,类朊病毒样的 hSOD1 聚集物的生长和传播可能是主要的致病机制,不仅在 hSOD1 转基因啮齿动物模型中,而且在人类 ALS 中也是如此。