Fujiwara Yu, Ito Kanae, Takamura Akito, Nagata Kaoru
Department of Internal Medicine, Musashino Red Cross Hospital, 1-26-1, Kyonancho, Musashino-shi, Tokyo, 1808610, Japan.
Department of Rheumatology and Collagen Disease, Musashino Red Cross Hospital, Tokyo, Japan.
J Med Case Rep. 2018 Oct 8;12(1):295. doi: 10.1186/s13256-018-1814-9.
TAFRO syndrome, which was first reported in 2010 in Japan, is a relatively rare disease characterized by thrombocytopenia, anasarca, fever, renal impairment, reticulin fibrosis, and organomegaly. Although this disease is considered similar to multicentric Castleman disease, some of the clinical features, such as thrombocytopenia, are different from typical cases of multicentric Castleman disease. In addition, the etiology of TAFRO syndrome remains unknown and controversial. There have only been a few cases of TAFRO syndrome complicated with adrenal gland lesions, and all of them have had hemorrhagic involvement.
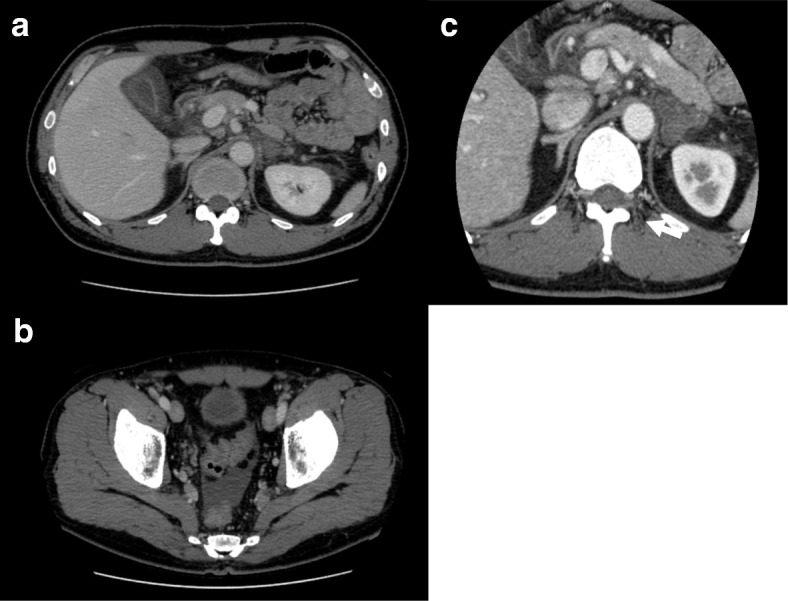
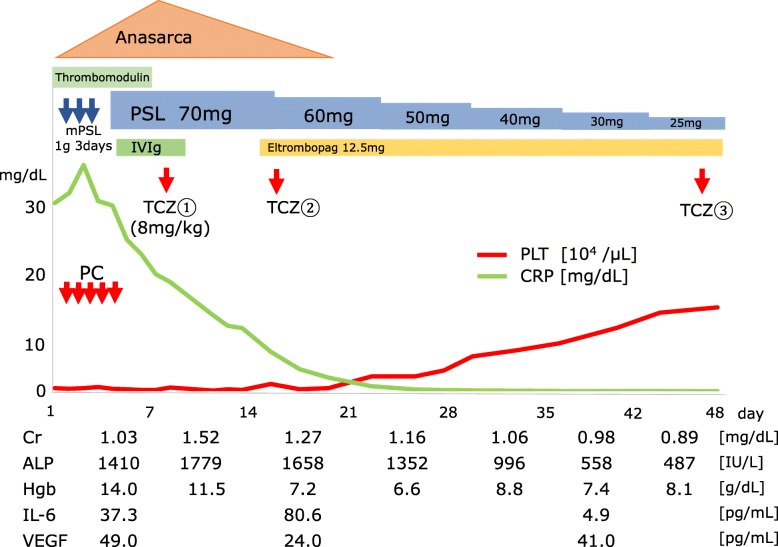
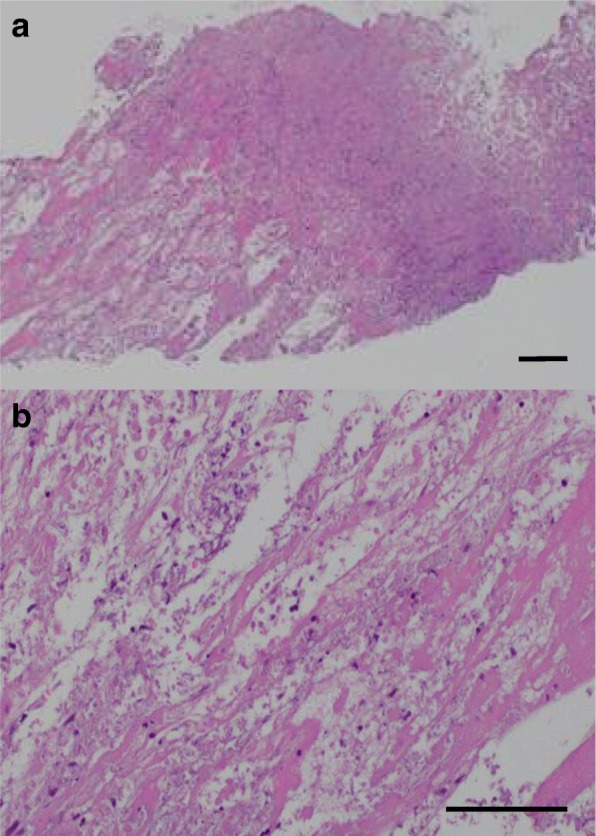
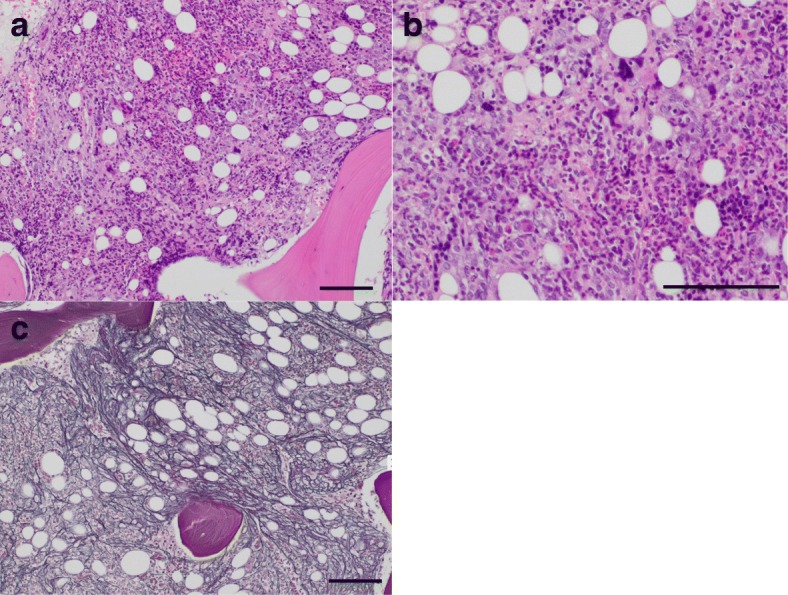
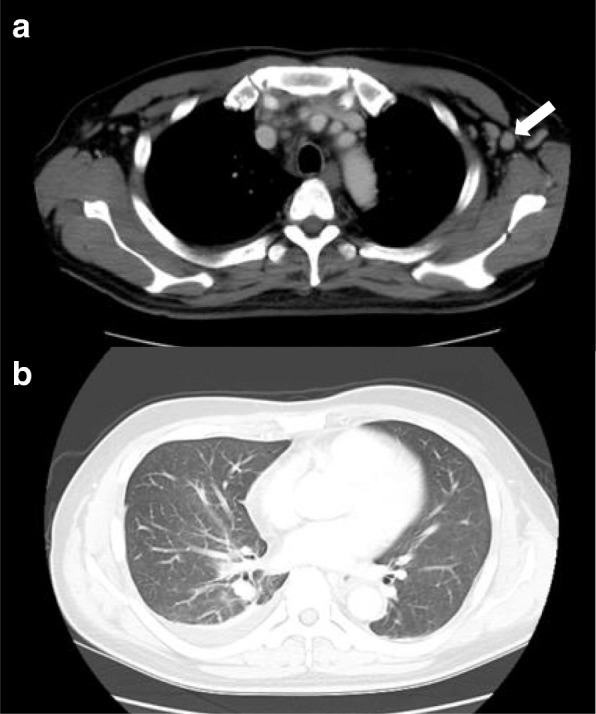
This report describes the case of a 46-year-old Asian man who presented with fever, epigastric pain, and back pain for 1 month. A computed tomographic scan revealed ascites, mild lymphadenopathy, and left adrenal necrosis without hemorrhage. A blood test showed thrombocytopenia, anemia, and elevated C-reactive protein, alkaline phosphatase, and creatinine levels. Based on the edema, severe thrombocytopenia, fever, reticulin myelofibrosis shown by bone marrow biopsy, mild lymphadenopathy, and progressive renal insufficiency, we diagnosed this patient as having TAFRO syndrome. He was successfully treated by immediate administration of glucocorticoids and tocilizumab.
There have been no previous reports of a case of TAFRO syndrome complicated with adrenal necrosis. Because the biopsy of the left adrenal gland revealed necrosis without any evidence of hemorrhage, we concluded that the unilateral adrenal necrosis in this case was caused by either ischemia from infarction or organomegaly itself under severe hypercytokinemia. This unusual clinical course is useful for further analysis of the etiology of TAFRO syndrome.
TAFRO综合征于2010年在日本首次报道,是一种相对罕见的疾病,其特征为血小板减少、全身性水肿、发热、肾功能损害、网状纤维组织增生及脏器肿大。尽管该疾病被认为与多中心Castleman病相似,但某些临床特征,如血小板减少,与典型的多中心Castleman病病例不同。此外,TAFRO综合征的病因仍不明且存在争议。仅有少数TAFRO综合征合并肾上腺病变的病例报道,且所有病例均有出血累及。
本报告描述了一名46岁亚洲男性的病例,该患者出现发热、上腹部疼痛和背痛1个月。计算机断层扫描显示有腹水、轻度淋巴结病及左侧肾上腺坏死但无出血。血液检查显示血小板减少、贫血,以及C反应蛋白、碱性磷酸酶和肌酐水平升高。基于水肿、严重血小板减少、发热、骨髓活检显示的网状纤维骨髓纤维化、轻度淋巴结病及进行性肾功能不全,我们诊断该患者患有TAFRO综合征。通过立即给予糖皮质激素和托珠单抗,他得到了成功治疗。
此前尚无TAFRO综合征合并肾上腺坏死病例的报道。由于左侧肾上腺活检显示坏死且无任何出血迹象,我们得出结论,该病例中的单侧肾上腺坏死是由梗死引起的缺血或严重高细胞因子血症下的脏器肿大本身所致。这种不寻常的临床病程有助于进一步分析TAFRO综合征的病因。