Ludwig Boltzmann Institute for Rare and Undiagnosed Diseases, Vienna, Austria.
CeMM Research Center for Molecular Medicine of the Austrian Academy of Sciences, Vienna, Austria.
Haematologica. 2019 Mar;104(3):609-621. doi: 10.3324/haematol.2018.194233. Epub 2018 Oct 11.
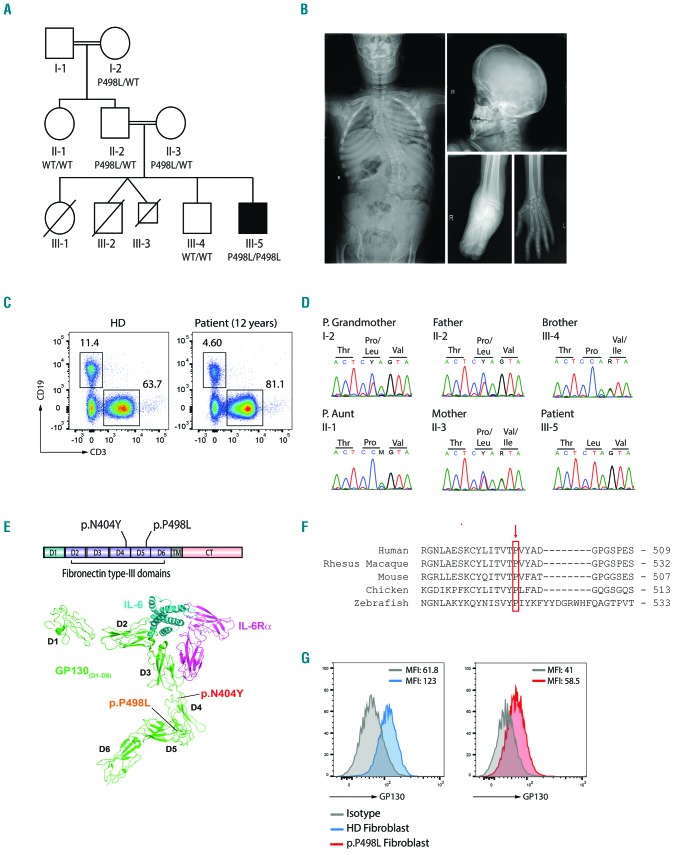
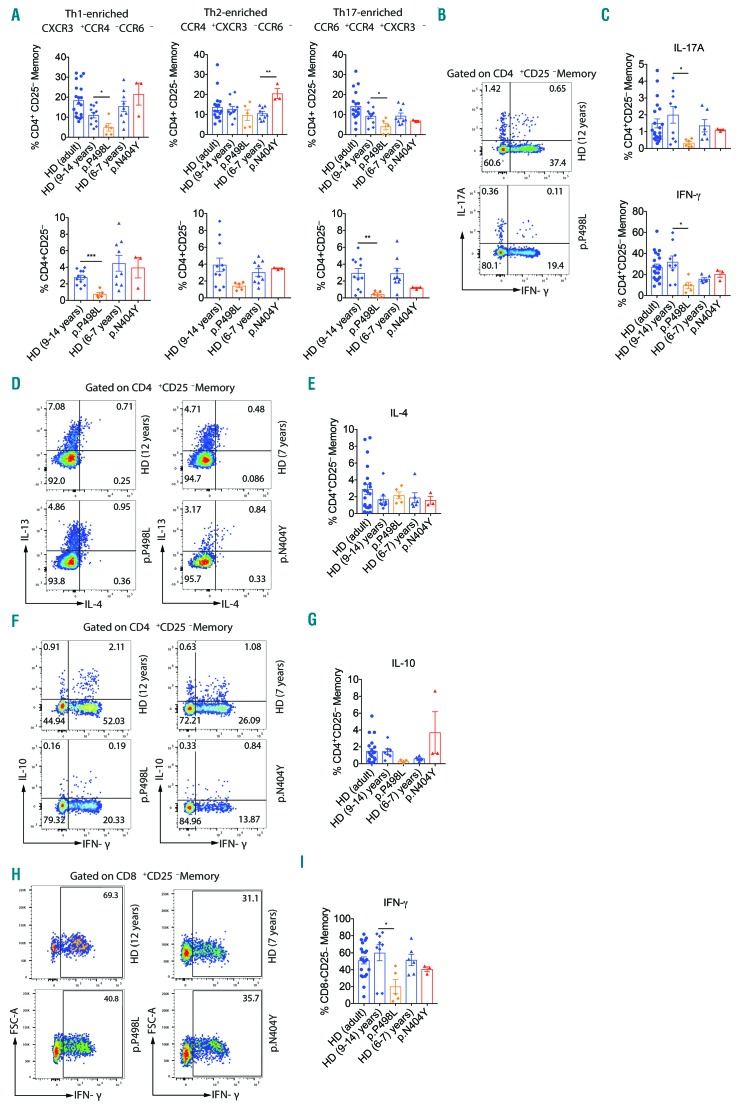
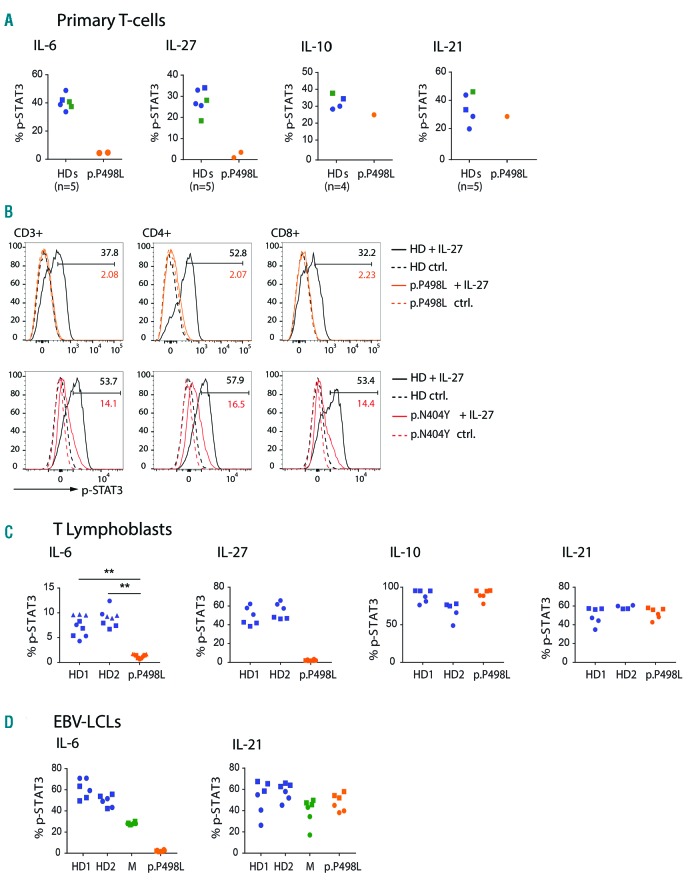
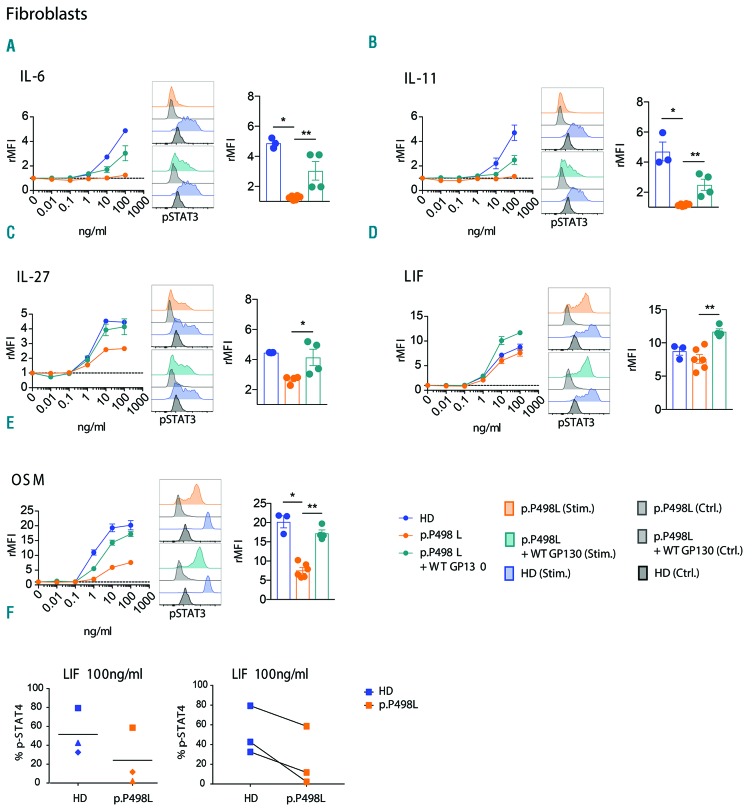
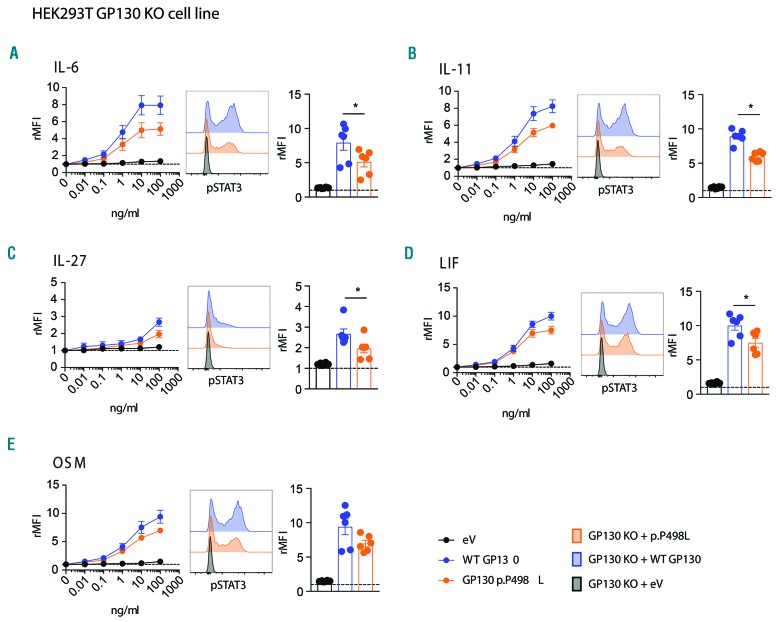
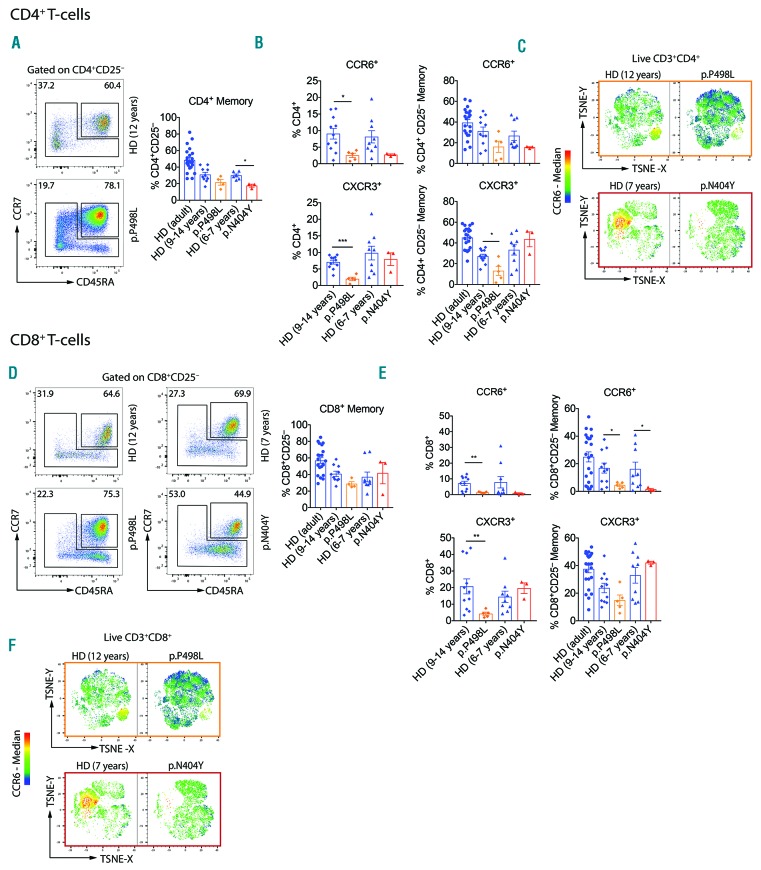
Hyper-IgE syndromes comprise a group of inborn errors of immunity. STAT3-deficient hyper-IgE syndrome is characterized by elevated serum IgE levels, recurrent infections and eczema, and characteristic skeletal anomalies. A loss-of-function biallelic mutation in encoding the GP130 receptor subunit (p.N404Y) has very recently been identified in a singleton patient (herein referred to as P) as a novel etiology of hyper-IgE syndrome. Here, we studied a patient with hyper-IgE syndrome caused by a novel homozygous mutation in (p.P498L; patient herein referred to as P) leading to abrogated GP130 signaling after stimulation with IL-6 and IL-27 in peripheral blood mononuclear cells as well as IL-6 and IL-11 in fibroblasts. Extending the initial identification of selective GP130 deficiency, we aimed to dissect the effects of aberrant cytokine signaling on T-helper cell differentiation in both patients. Our results reveal the importance of IL-6 signaling for the development of CCR6-expressing memory CD4 T cells (including T-helper 17-enriched subsets) and non-conventional CD8T cells which were reduced in both patients. Downstream functional analysis of the GP130 mutants (p.N404Y and p.P498L) have shown differences in response to IL-27, with the p.P498L mutation having a more severe effect that is reflected by reduced T-helper 1 cells in this patient (P) only. Collectively, our data suggest that characteristic features of GP130-deficient hyper-IgE syndrome phenotype are IL-6 and IL-11 dominated, and indicate selective roles of aberrant IL-6 and IL-27 signaling on the differentiation of T-cell subsets.
高免疫球蛋白 E 综合征包括一组先天性免疫缺陷。STAT3 缺陷型高免疫球蛋白 E 综合征的特征是血清 IgE 水平升高、反复感染和湿疹以及特征性骨骼异常。最近在一个单例患者(以下简称 P)中发现编码 GP130 受体亚基的 (p.N404Y)的功能丧失性双等位基因突变,这是高免疫球蛋白 E 综合征的一种新病因。在这里,我们研究了一名高免疫球蛋白 E 综合征患者,该患者因 (p.P498L;以下简称 P)中的新型纯合突变导致外周血单个核细胞中 IL-6 和 IL-27 以及成纤维细胞中 IL-6 和 IL-11 刺激后 GP130 信号转导被阻断。扩展最初确定的选择性 GP130 缺乏症,我们旨在剖析异常细胞因子信号对这两个患者的辅助性 T 细胞分化的影响。我们的结果揭示了 IL-6 信号在 CCR6 表达的记忆性 CD4 T 细胞(包括富含辅助性 T 细胞 17 的亚群)和非传统 CD8T 细胞发育中的重要性,这两种细胞在两个患者中均减少。对 GP130 突变体(p.N404Y 和 p.P498L)的下游功能分析表明,对 IL-27 的反应存在差异,p.P498L 突变的影响更为严重,这在该患者(P)中仅表现为辅助性 T 细胞 1 减少。总的来说,我们的数据表明,GP130 缺陷型高免疫球蛋白 E 综合征表型的特征是由 IL-6 和 IL-11 主导的,并表明异常的 IL-6 和 IL-27 信号对 T 细胞亚群分化具有选择性作用。