Ortega-Muñoz Mariano, Rodríguez-Serrano Fernando, De Los Reyes-Berbel Eduardo, Mut-Salud Nuria, Hernández-Mateo Fernando, Rodríguez-López Andrea, Garrido José M, López-Jaramillo F Javier, Santoyo-González Francisco
Department of Organic Chemistry, Faculty of Sciences, Department of Organic Chemistry, Biotechnology Institute, Institute of Biopathology and Regenerative Medicine (IBIMER), and Department of Human Anatomy and Embryology, University of Granada, 18071 Granada, Spain.
Biosanitary Research Institute of Granada (ibs.GRANADA), 18071 Granada, Spain.
ACS Omega. 2018 Sep 30;3(9):11455-11468. doi: 10.1021/acsomega.8b01034. Epub 2018 Sep 20.
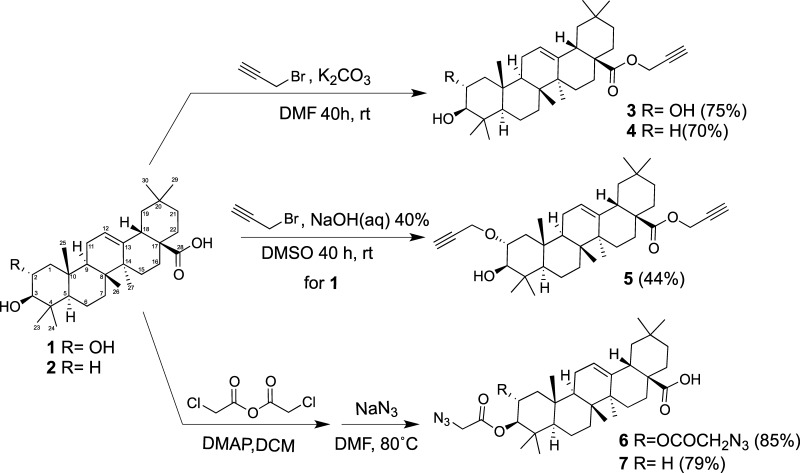
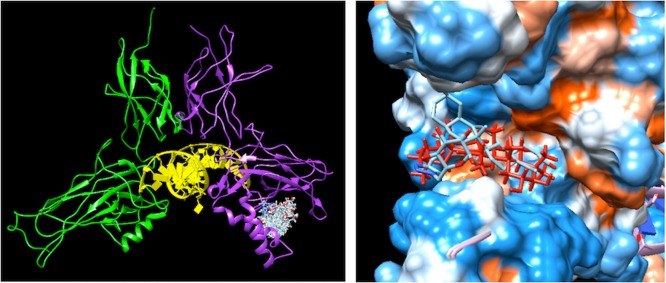
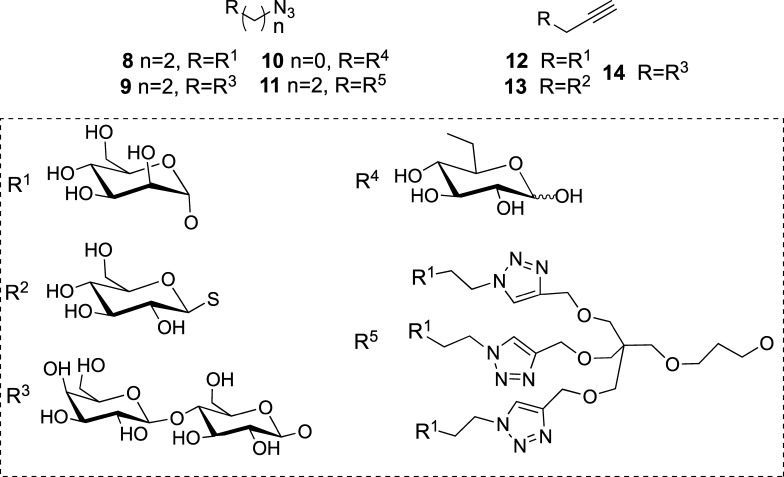
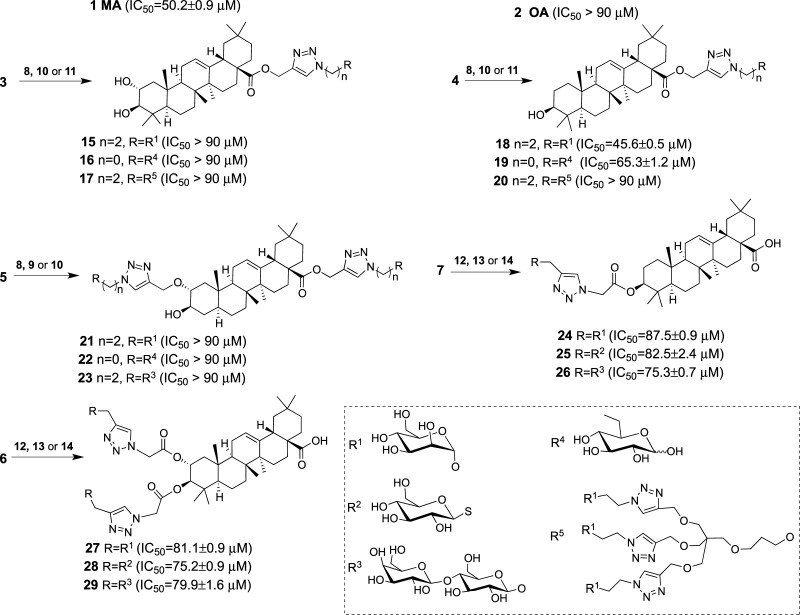
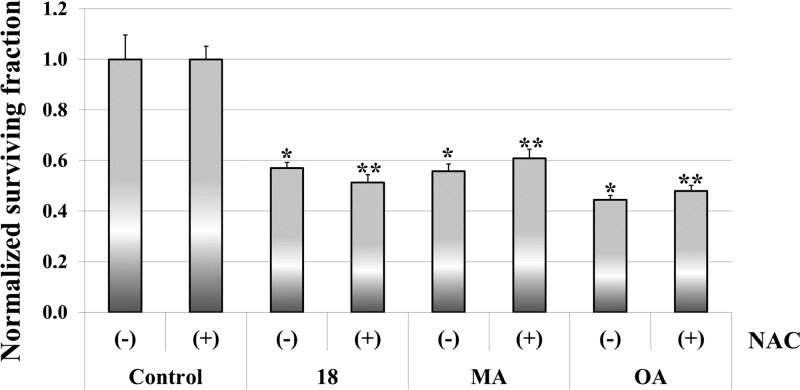
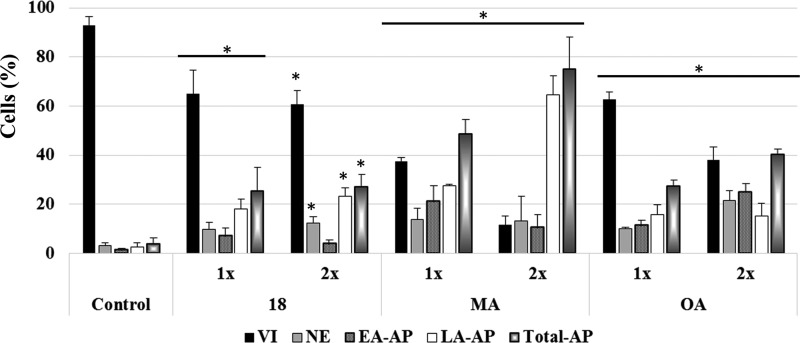
Saponins are potential wide-spectrum antitumor drugs, and copper(I) catalyzed azide-alkyne 1,3-dipolar cycloaddition is a suitable approach to synthesizing saponin-like compounds by regioselective glycosylation of the C2/C3 hydroxyl and C28 carboxylic groups of triterpene aglycones maslinic acid (MA) and oleanolic acid (OA). Biological studies on the T-84 human colon carcinoma cell line support the role of the hydroxyl groups at C2/C3, the influence of the aglycone, and the bulky nature of the substituents in C28. OA bearing a α-d-mannose moiety at C28 (compound ) focused our interest because the estimated inhibitory concentration 50 was similar to that reported for ginsenoside Rh2 against colon cancer cells and it inhibits the G-S phase transition affecting the cell viability and apoptosis. Considering that triterpenoids from natural sources have been identified as inhibitors of nuclear factor kappa-light-chain-enhancer of activated B cell (NF-κB) signaling, docking studies were conducted to evaluate whether NF-κB may be a potential target. Results are consistent with the biological study and predict a similar binding mode of MA and compound to the p52 subunit from NF-κB but not for OA. The fact that the binding site is shared by the NF-κB inhibitor 6,6-dimethyl-2-(phenylimino)-6,7-dihydrobenzo[][1,3]oxathiol-4(5)-one supports the result and points to NF-κB as a potential target of both MA and compound .
皂苷是潜在的广谱抗肿瘤药物,铜(I)催化的叠氮化物-炔烃1,3-偶极环加成反应是通过对三萜苷元山楂酸(MA)和齐墩果酸(OA)的C2/C3羟基和C28羧基进行区域选择性糖基化来合成皂苷类化合物的合适方法。对T-84人结肠癌细胞系的生物学研究支持了C2/C3羟基的作用、苷元的影响以及C28取代基的庞大性质。在C28带有α-d-甘露糖部分的OA(化合物 )引起了我们的兴趣,因为估计的半数抑制浓度与报道的人参皂苷Rh2对结肠癌细胞的抑制浓度相似,并且它抑制G-S期转变,影响细胞活力和凋亡。鉴于天然来源的三萜类化合物已被鉴定为活化B细胞核因子κB轻链增强子(NF-κB)信号的抑制剂,进行了对接研究以评估NF-κB是否可能是潜在靶点。结果与生物学研究一致,并预测MA和化合物 与NF-κB的p52亚基具有相似的结合模式,但OA并非如此。NF-κB抑制剂6,6-二甲基-2-(苯基亚氨基)-6,7-二氢苯并[[1,3]氧杂硫醇-4(5)-酮共享结合位点这一事实支持了该结果,并表明NF-κB是MA和化合物 的潜在靶点。