Haider Saad, Black Michael B, Parks Bethany B, Foley Briana, Wetmore Barbara A, Andersen Melvin E, Clewell Rebecca A, Mansouri Kamel, McMullen Patrick D
ScitoVation, Research Triangle Park, NC, United States.
Front Pharmacol. 2018 Oct 2;9:1072. doi: 10.3389/fphar.2018.01072. eCollection 2018.
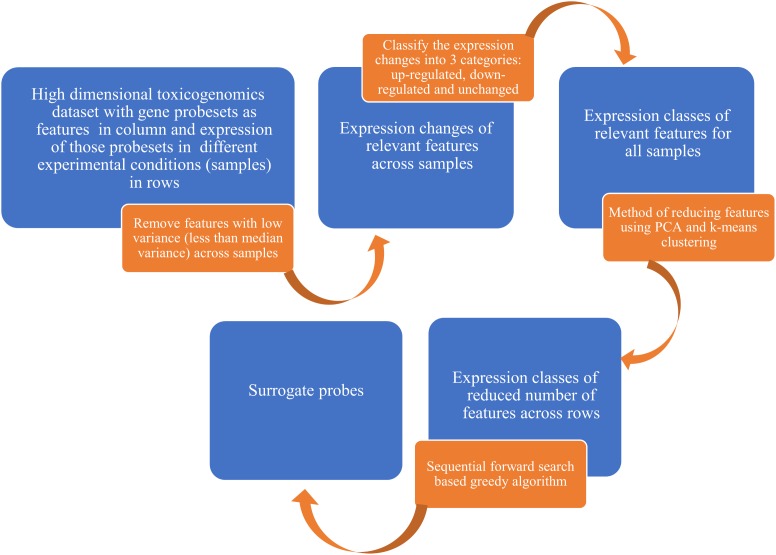
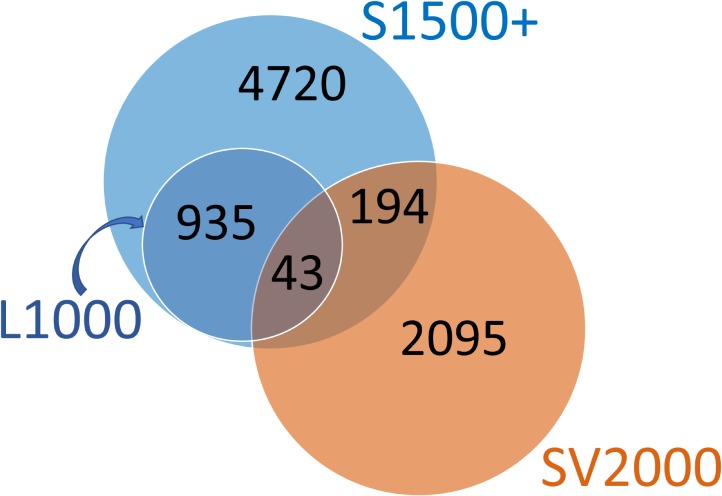
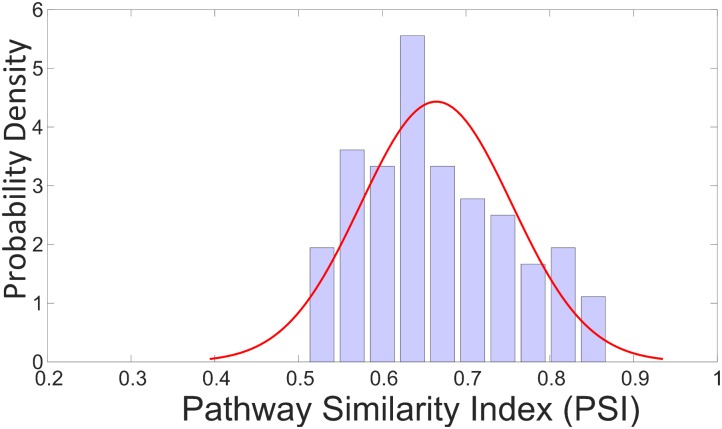
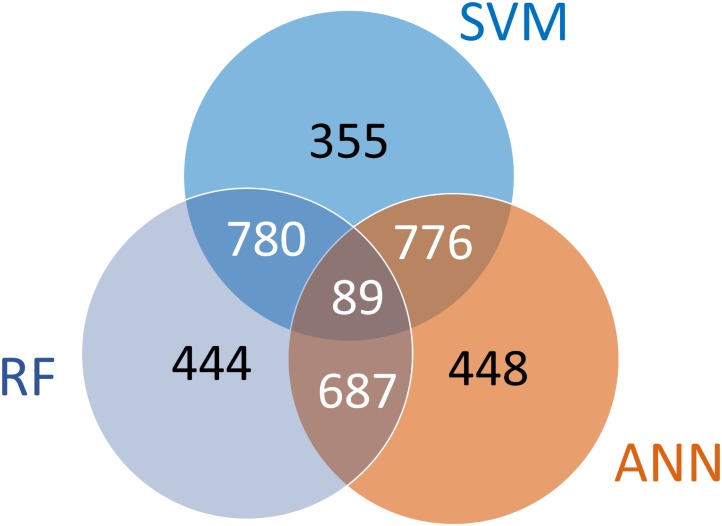
Efficient high-throughput transcriptomics (HTT) tools promise inexpensive, rapid assessment of possible biological consequences of human and environmental exposures to tens of thousands of chemicals in commerce. HTT systems have used relatively small sets of gene expression measurements coupled with mathematical prediction methods to estimate genome-wide gene expression and are often trained and validated using pharmaceutical compounds. It is unclear whether these training sets are suitable for general toxicity testing applications and the more diverse chemical space represented by commercial chemicals and environmental contaminants. In this work, we built predictive computational models that inferred whole genome transcriptional profiles from a smaller sample of surrogate genes. The model was trained and validated using a large scale toxicogenomics database with gene expression data from exposure to heterogeneous chemicals from a wide range of classes (the Open TG-GATEs data base). The method of predictor selection was designed to allow high fidelity gene prediction from any pre-existing gene expression data set, regardless of animal species or data measurement platform. Predictive qualitative models were developed with this TG-GATES data that contained gene expression data of human primary hepatocytes with over 941 samples covering 158 compounds. A sequential forward search-based greedy algorithm, combining different fitting approaches and machine learning techniques, was used to find an optimal set of surrogate genes that predicted differential expression changes of the remaining genome. We then used pathway enrichment of up-regulated and down-regulated genes to assess the ability of a limited gene set to determine relevant patterns of tissue response. In addition, we compared prediction performance using the surrogate genes found from our greedy algorithm (referred to as the SV2000) with the landmark genes provided by existing technologies such as L1000 (Genometry) and S1500 (Tox21), finding better predictive performance for the SV2000. The ability of these predictive algorithms to predict pathway level responses is a positive step toward incorporating mode of action (MOA) analysis into the high throughput prioritization and testing of the large number of chemicals in need of safety evaluation.
高效的高通量转录组学(HTT)工具有望以低成本、快速评估人类和环境接触商业中数以万计化学物质可能产生的生物学后果。HTT系统使用相对较少的基因表达测量集,并结合数学预测方法来估计全基因组基因表达,且通常使用药物化合物进行训练和验证。目前尚不清楚这些训练集是否适用于一般毒性测试应用以及商业化学品和环境污染物所代表的更多样化的化学空间。在这项工作中,我们构建了预测性计算模型,该模型可从较小的替代基因样本推断全基因组转录谱。该模型使用一个大规模毒理基因组学数据库进行训练和验证,该数据库包含来自广泛类别的异质化学物质暴露的基因表达数据(开放毒理基因组学数据库)。预测器选择方法的设计旨在允许从任何现有基因表达数据集中进行高保真基因预测,而不论动物物种或数据测量平台如何。利用该包含158种化合物的941多个样本的人类原代肝细胞基因表达数据的TG-GATES数据开发了预测性定性模型。一种基于顺序向前搜索的贪婪算法,结合不同的拟合方法和机器学习技术,用于找到一组最佳的替代基因,以预测其余基因组的差异表达变化。然后,我们使用上调和下调基因的通路富集来评估有限基因集确定组织反应相关模式的能力。此外,我们将使用贪婪算法找到的替代基因(称为SV2000)的预测性能与现有技术(如L1000(Genometry)和S1500(Tox21))提供的标志性基因进行了比较,发现SV2000具有更好的预测性能。这些预测算法预测通路水平反应的能力是朝着将作用模式(MOA)分析纳入大量需要安全评估的化学物质的高通量优先级排序和测试迈出的积极一步。