Mav Deepak, Shah Ruchir R, Howard Brian E, Auerbach Scott S, Bushel Pierre R, Collins Jennifer B, Gerhold David L, Judson Richard S, Karmaus Agnes L, Maull Elizabeth A, Mendrick Donna L, Merrick B Alex, Sipes Nisha S, Svoboda Daniel, Paules Richard S
SciOme LLC, Research Triangle Park, North Carolina, United States of America.
Division of the National Toxicology Program, National Institute of Environmental Health Sciences, National Institutes of Health, Research Triangle Park, North Carolina, United States of America.
PLoS One. 2018 Feb 20;13(2):e0191105. doi: 10.1371/journal.pone.0191105. eCollection 2018.
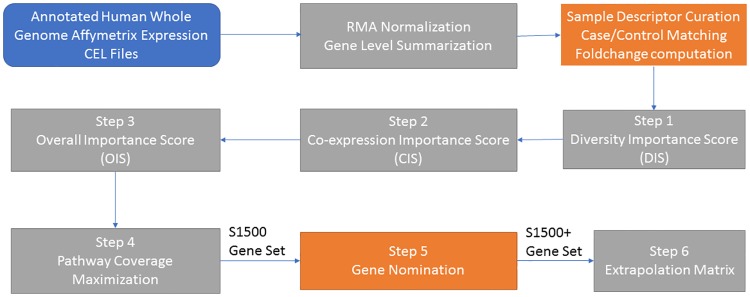
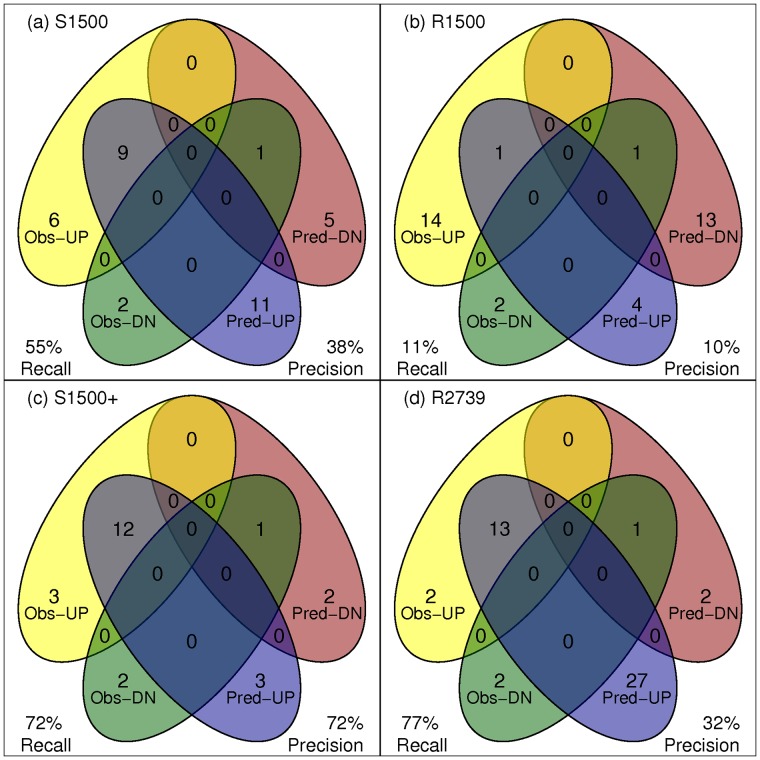
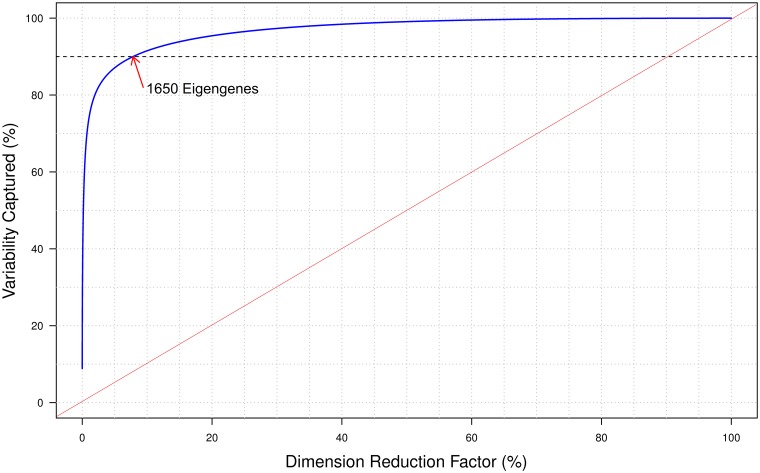
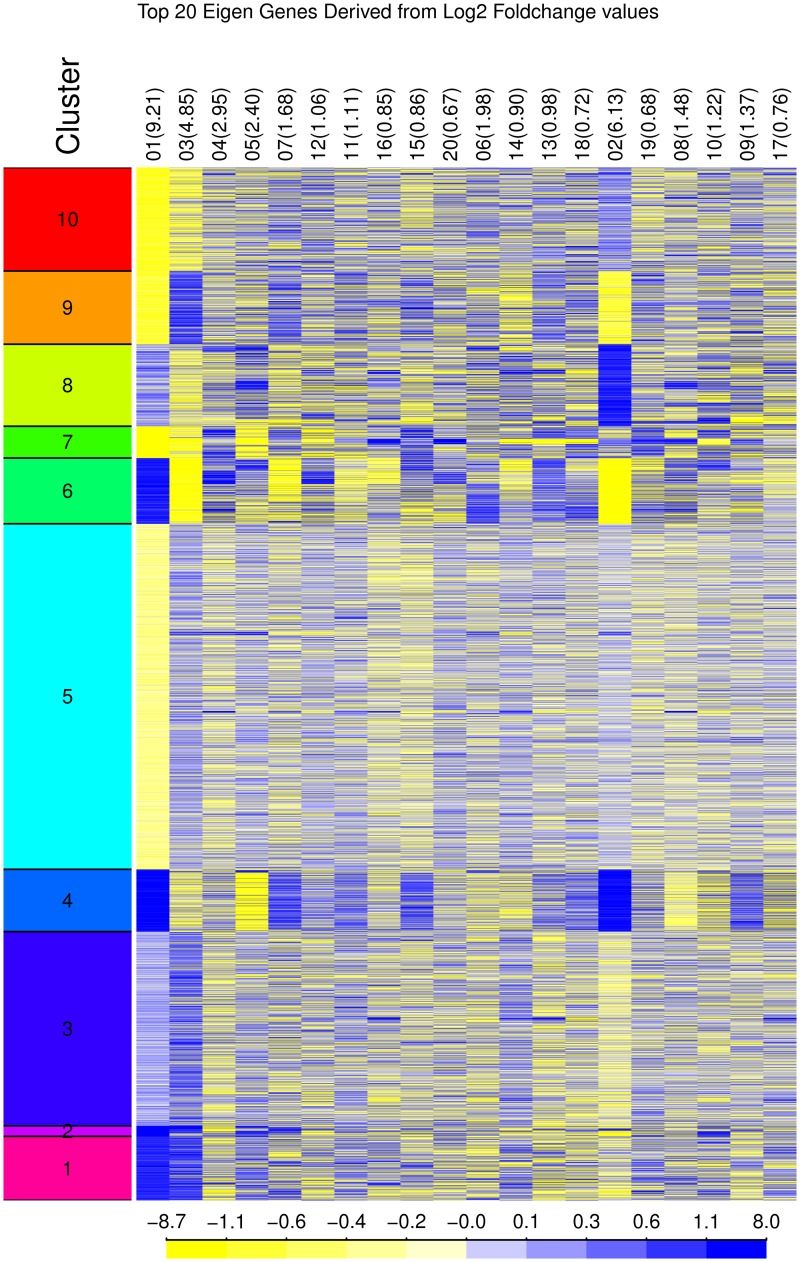
Changes in gene expression can help reveal the mechanisms of disease processes and the mode of action for toxicities and adverse effects on cellular responses induced by exposures to chemicals, drugs and environment agents. The U.S. Tox21 Federal collaboration, which currently quantifies the biological effects of nearly 10,000 chemicals via quantitative high-throughput screening(qHTS) in in vitro model systems, is now making an effort to incorporate gene expression profiling into the existing battery of assays. Whole transcriptome analyses performed on large numbers of samples using microarrays or RNA-Seq is currently cost-prohibitive. Accordingly, the Tox21 Program is pursuing a high-throughput transcriptomics (HTT) method that focuses on the targeted detection of gene expression for a carefully selected subset of the transcriptome that potentially can reduce the cost by a factor of 10-fold, allowing for the analysis of larger numbers of samples. To identify the optimal transcriptome subset, genes were sought that are (1) representative of the highly diverse biological space, (2) capable of serving as a proxy for expression changes in unmeasured genes, and (3) sufficient to provide coverage of well described biological pathways. A hybrid method for gene selection is presented herein that combines data-driven and knowledge-driven concepts into one cohesive method. Our approach is modular, applicable to any species, and facilitates a robust, quantitative evaluation of performance. In particular, we were able to perform gene selection such that the resulting set of "sentinel genes" adequately represents all known canonical pathways from Molecular Signature Database (MSigDB v4.0) and can be used to infer expression changes for the remainder of the transcriptome. The resulting computational model allowed us to choose a purely data-driven subset of 1500 sentinel genes, referred to as the S1500 set, which was then augmented using a knowledge-driven selection of additional genes to create the final S1500+ gene set. Our results indicate that the sentinel genes selected can be used to accurately predict pathway perturbations and biological relationships for samples under study.
基因表达的变化有助于揭示疾病过程的机制以及化学物质、药物和环境因子暴露对细胞反应产生毒性和不良反应的作用模式。美国毒物21联邦合作项目目前通过体外模型系统中的定量高通量筛选(qHTS)对近10000种化学物质的生物学效应进行量化,现正努力将基因表达谱分析纳入现有的检测方法组中。使用微阵列或RNA测序对大量样本进行全转录组分析目前成本过高。因此,毒物21项目正在寻求一种高通量转录组学(HTT)方法,该方法专注于对转录组中精心挑选的子集进行基因表达的靶向检测,这有可能将成本降低10倍,从而能够分析更多的样本。为了确定最佳的转录组子集,寻找了以下几类基因:(1)代表高度多样化生物空间的基因;(2)能够替代未测量基因表达变化的基因;(3)足以覆盖充分描述的生物途径的基因。本文提出了一种基因选择的混合方法,该方法将数据驱动和知识驱动的概念整合为一种连贯的方法。我们的方法是模块化的,适用于任何物种,并有助于对性能进行稳健的定量评估。特别是,我们能够进行基因选择,使得所得的“哨兵基因”集能够充分代表分子特征数据库(MSigDB v4.0)中的所有已知经典途径,并可用于推断转录组其余部分的表达变化。所得的计算模型使我们能够选择一个由1500个哨兵基因组成的纯数据驱动子集,称为S1500集,然后通过知识驱动选择额外的基因对其进行扩充,以创建最终的S1500+基因集。我们的结果表明,所选的哨兵基因可用于准确预测所研究样本的途径扰动和生物学关系。