APHP, Reference Center for Rare Disorders of the Calcium and Phosphate Metabolism, Filière OSCAR, Bicêtre Paris Sud Hospital, Le Kremlin Bicêtre, France.
APHP, Endocrinology and Diabetes for Children, Bicêtre Paris Sud Hospital, Le Kremlin Bicêtre, France.
J Bone Miner Res. 2019 Mar;34(3):490-496. doi: 10.1002/jbmr.3614. Epub 2018 Nov 20.
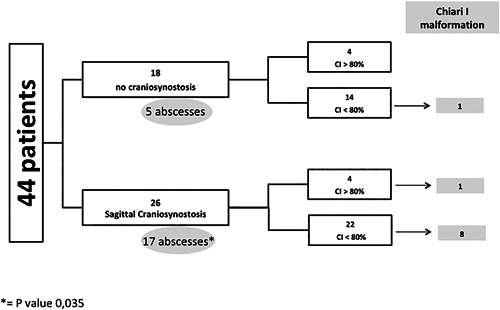
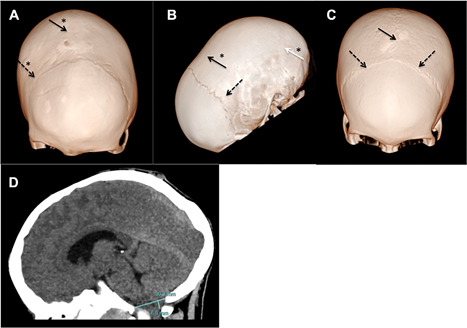
X-linked hypophosphatemic rickets (XLHR) represents the most common form of genetic hypophosphatemia and causes rickets and osteomalacia in children because of increased FGF23 secretion and renal phosphate wasting. Even though cranial vault and craniovertebral anomalies of potential neurosurgical interest, namely early closure of the cranial sutures and Chiari type I malformation, have been observed in children with XLHR, their actual incidence and characteristics are not established. The aims of this study were to analyze the incidence of cranial and cervico-occipital junction (COJ) anomalies in children with XLHR and describe its features. This is a retrospective study of CT scans of the head and skull in 44 XLHR children followed at the French Reference Center for Rare Diseases of the Calcium and Phosphate Metabolism. Forty-four children with XLHR, 15 boys and 29 girls, aged 8.7 ± 3.9 years at time of CT scan, were studied. We found that 59% of XLHR children had a complete or partial fusion of the sagittal suture and 25% of XLHR children showed protrusion of the cerebellar tonsils. A history of dental abscesses was associated with craniosynostosis, and craniosynostosis was associated with abnormal descent of cerebellar tonsils. Only 2 patients showed neurologic symptoms. Four of 44 patients (9%) required neurosurgery. This study highlights that sagittal suture fusion and Chiari type I malformation are frequent complications of XLHR. The incidence of sagittal synostosis in XLHR is actually extremely high and was probably underestimated so far. Chiari type I malformation is also frequent. Because diagnosis of craniovertebral anomalies can be underestimated on a purely clinical basis, radiological studies should be considered in XLHR children if a proper diagnosis is warranted. © 2018 The Authors. Journal of Bone and Mineral Research Published by Wiley Periodicals, Inc.
X 连锁低磷血症性佝偻病(XLHR)是最常见的遗传性低磷血症形式,由于 FGF23 分泌增加和肾脏磷酸盐丢失,导致儿童佝偻病和骨软化症。尽管在 XLHR 儿童中已经观察到具有潜在神经外科意义的颅顶和颅颈交界区(COJ)异常,即颅缝早期闭合和 Chiari Ⅰ型畸形,但它们的实际发生率和特征尚未确定。本研究旨在分析 XLHR 儿童颅和颈椎-枕骨接合处(COJ)异常的发生率并描述其特征。这是对法国钙和磷酸盐代谢罕见疾病参考中心随访的 44 例 XLHR 儿童头颅和颅骨 CT 扫描的回顾性研究。44 例 XLHR 儿童(15 名男孩和 29 名女孩),在进行 CT 扫描时的年龄为 8.7±3.9 岁,我们发现 59%的 XLHR 儿童存在矢状缝完全或部分融合,25%的 XLHR 儿童存在小脑扁桃体突出。牙脓肿病史与颅缝早闭相关,而颅缝早闭与小脑扁桃体异常下降相关。仅有 2 例患者出现神经系统症状。44 例患者中有 4 例(9%)需要神经外科手术。本研究强调矢状缝融合和 Chiari Ⅰ型畸形是 XLHR 的常见并发症。XLHR 中矢状缝早闭的发生率实际上极高,迄今为止可能被低估。Chiari Ⅰ型畸形也很常见。由于颅颈交界区异常仅凭临床诊断可能被低估,如果需要明确诊断,应考虑对 XLHR 儿童进行影像学研究。