Department of Computer Science and Engineering, Pennsylvania State University, University Park, PA, 16802, USA.
Department of Biology, Pennsylvania State University, University Park, PA, 16802, USA.
Nat Commun. 2018 Nov 2;9(1):4601. doi: 10.1038/s41467-018-06910-x.
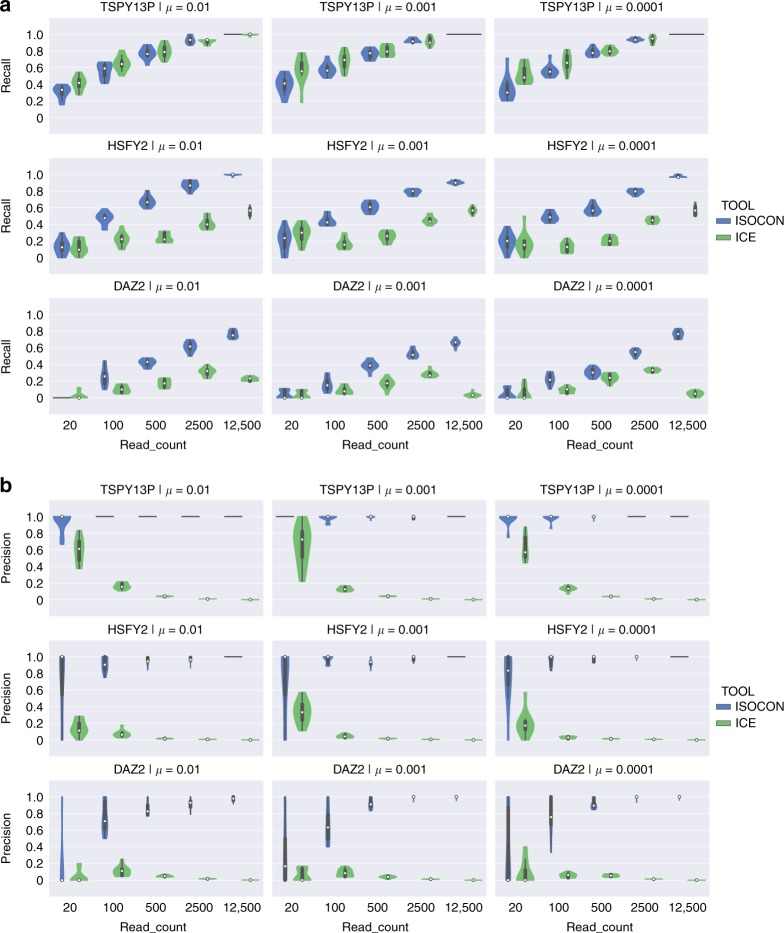
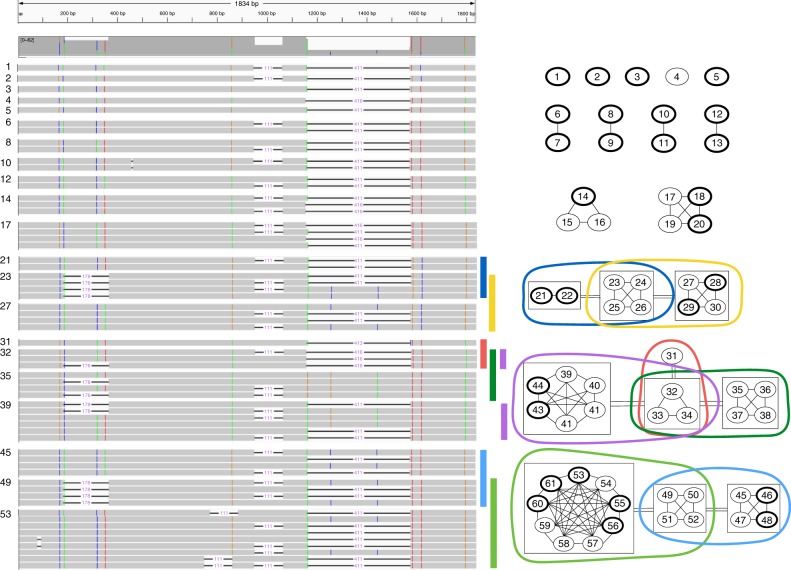
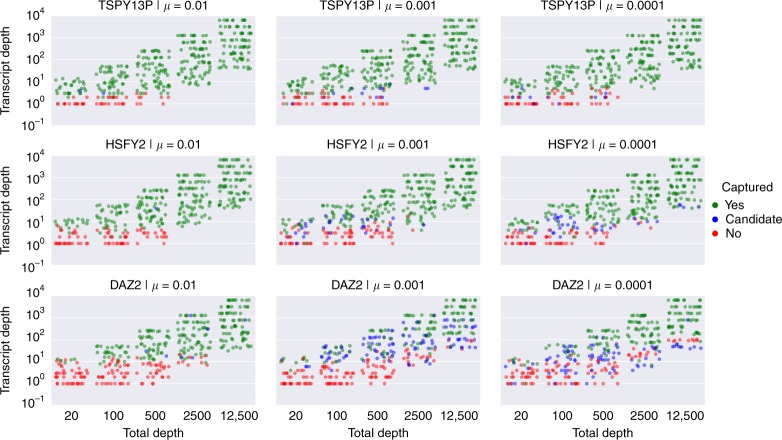
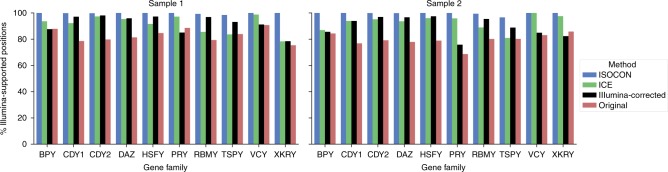
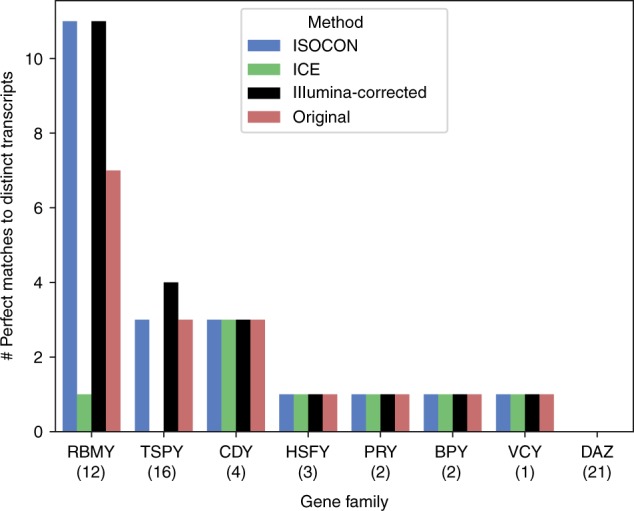
A significant portion of genes in vertebrate genomes belongs to multigene families, with each family containing several gene copies whose presence/absence, as well as isoform structure, can be highly variable across individuals. Existing de novo techniques for assaying the sequences of such highly-similar gene families fall short of reconstructing end-to-end transcripts with nucleotide-level precision or assigning alternatively spliced transcripts to their respective gene copies. We present IsoCon, a high-precision method using long PacBio Iso-Seq reads to tackle this challenge. We apply IsoCon to nine Y chromosome ampliconic gene families and show that it outperforms existing methods on both experimental and simulated data. IsoCon has allowed us to detect an unprecedented number of novel isoforms and has opened the door for unraveling the structure of many multigene families and gaining a deeper understanding of genome evolution and human diseases.
脊椎动物基因组中的很大一部分基因属于多基因家族,每个家族包含几个基因副本,其存在/缺失以及同工型结构在个体之间可能有很大差异。现有的从头分析这些高度相似的基因家族序列的技术无法精确重建核苷酸水平的全长转录本,也无法将选择性剪接的转录本分配给各自的基因副本。我们提出了 IsoCon,这是一种使用长 PacBio Iso-Seq reads 来解决这一挑战的高精度方法。我们将 IsoCon 应用于九个 Y 染色体扩增基因家族,并表明它在实验和模拟数据上都优于现有方法。IsoCon 使我们能够检测到前所未有的大量新同工型,并为揭示许多多基因家族的结构以及更深入地了解基因组进化和人类疾病打开了大门。