Cuneo Kyle C, Mehta Ranjit K, Kurapati Himabindu, Thomas Dafydd G, Lawrence Theodore S, Nyati Mukesh K
University of Michigan Medical School, Department of Radiation Oncology, Ann Arbor, MI, USA.
University of Michigan Medical School, Department of Radiation Oncology, Ann Arbor, MI, USA.
Transl Oncol. 2019 Feb;12(2):209-216. doi: 10.1016/j.tranon.2018.10.005. Epub 2018 Nov 6.
C-Met plays important roles in treatment resistance, tumor invasion, and metastasis. In this study, we used a small molecule inhibitor of c-Met, crizotinib, in cetuximab-resistant, mutant KRAS-driven colorectal cancer cell lines and assessed radiosensitization.
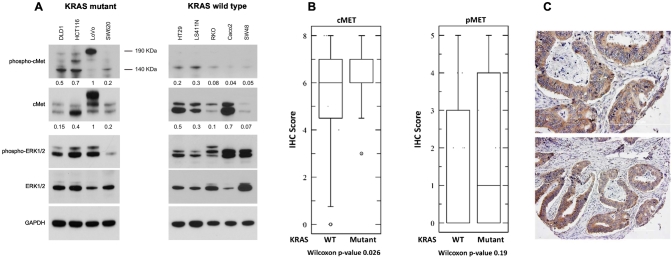
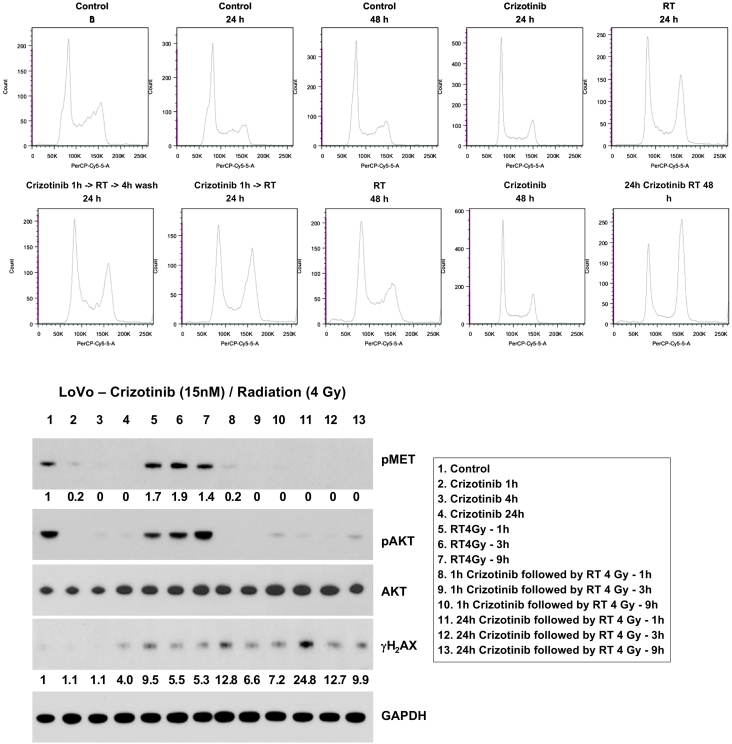
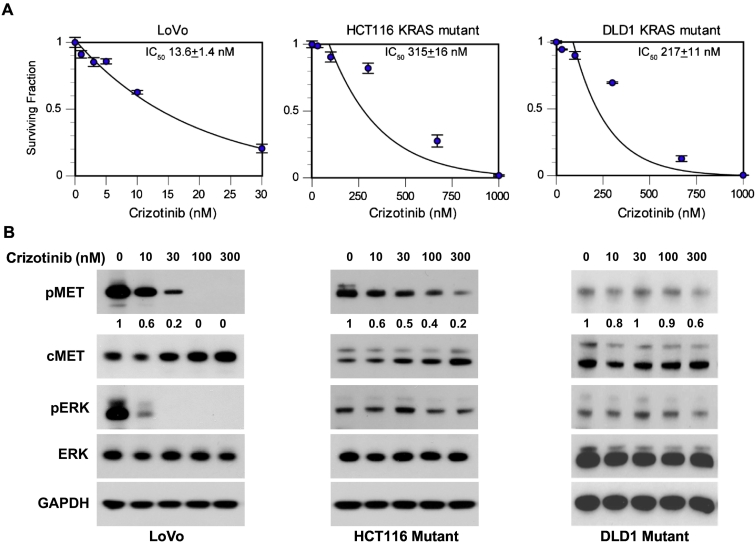
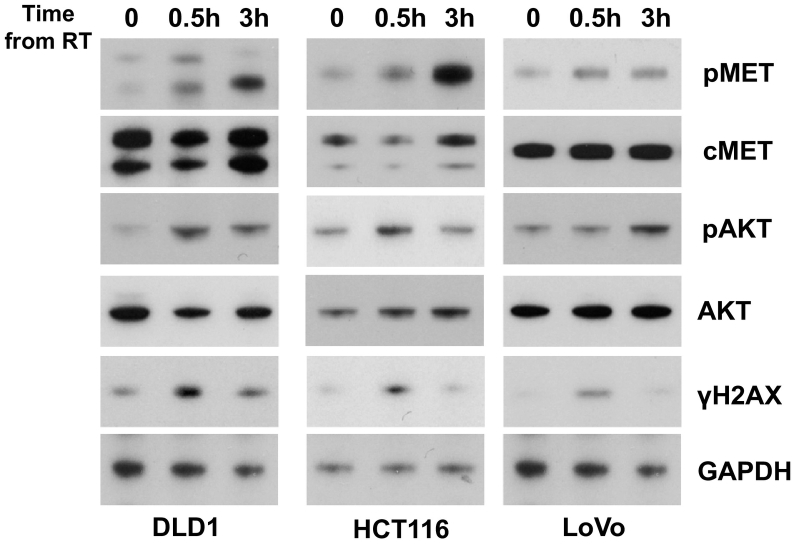
A tissue microarray containing colorectal tumors was used to study the relationship between KRAS mutations and c-Met expression. For in vivo studies, we used the KRAS mutant cell lines HCT116, DLD1, and LoVo. Colony formation assays were performed to assess the effects of crizotinib and cetuximab. Immunoblot analysis was used to determine the effect of crizotinib on c-Met and downstream pathways and DNA damage response. We then selected noncytotoxic doses of crizotinib to assess clonogenic survival with radiation. To study potential mechanisms of radiosensitization, cell cycle analysis was performed using flow cytometry.
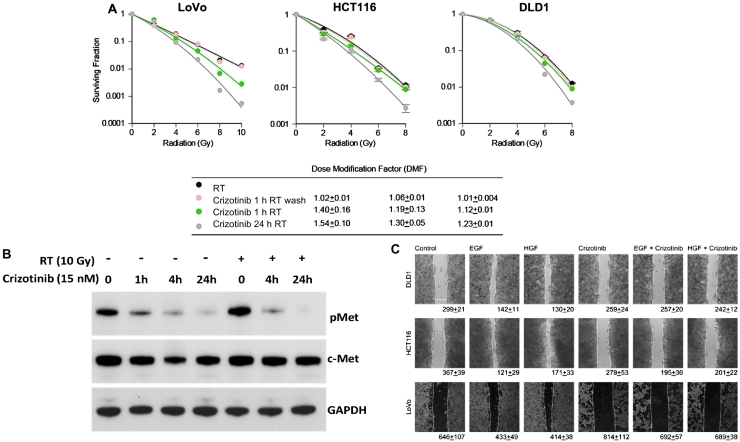
Analysis of the tissue microarray revealed that KRAS mutant tumors had active c-Met signaling. KRAS mutant cell lines LoVo, HCT116, and DLD1 were resistant to cetuximab but sensitive to crizotinib. Pretreatment with crizotinib for 24 hours radiosensitized LoVo, DLD1, and HCT116 cell lines with enhancement ratios of 1.54, 1.23, and 1.30, respectively. Immunoblot analysis showed that crizotinib blocked radiation-induced c-Met phosphorylation and attenuated downstream signaling pathways. Cell cycle analysis revealed minimal G1 arrest with crizotinib. Additionally, crizotinib completely blocked HGF induced cell migration.
Inhibition of c-Met with crizotinib effectively sensitizes cetuximab-resistant KRAS mutant colorectal cancer cell lines to radiation. Crizotinib has the potential to improve outcomes in locally advanced rectal cancer patients undergoing chemoradiation.
C-Met在治疗耐药、肿瘤侵袭和转移中发挥重要作用。在本研究中,我们在西妥昔单抗耐药、KRAS突变驱动的结直肠癌细胞系中使用了C-Met的小分子抑制剂克唑替尼,并评估了放射增敏作用。
使用包含结直肠肿瘤的组织微阵列研究KRAS突变与C-Met表达之间的关系。对于体内研究,我们使用了KRAS突变细胞系HCT116、DLD1和LoVo。进行集落形成试验以评估克唑替尼和西妥昔单抗的作用。免疫印迹分析用于确定克唑替尼对C-Met及其下游通路和DNA损伤反应的影响。然后我们选择了克唑替尼的非细胞毒性剂量来评估放疗后的克隆形成存活率。为了研究放射增敏的潜在机制,使用流式细胞术进行细胞周期分析。
组织微阵列分析显示KRAS突变肿瘤具有活跃的C-Met信号传导。KRAS突变细胞系LoVo、HCT116和DLD1对西妥昔单抗耐药,但对克唑替尼敏感。用克唑替尼预处理24小时可使LoVo、DLD1和HCT116细胞系放射增敏,增强比分别为1.54、1.23和1.30。免疫印迹分析表明,克唑替尼可阻断辐射诱导的C-Met磷酸化并减弱下游信号通路。细胞周期分析显示克唑替尼对G1期阻滞作用极小。此外,克唑替尼完全阻断了HGF诱导的细胞迁移。
用克唑替尼抑制C-Met可有效使西妥昔单抗耐药的KRAS突变结直肠癌细胞系对放疗敏感。克唑替尼有潜力改善接受放化疗的局部晚期直肠癌患者的治疗效果。