Max Planck Institute for Molecular Genetics, 14195 Berlin, Germany, Ihnestraße 63-73, Berlin, 14195, Germany.
BMC Biol. 2018 Nov 15;16(1):138. doi: 10.1186/s12915-018-0585-5.
Characterizing recurring sequence patterns in human promoters has been a challenging undertaking even nowadays where a near-complete overview of promoters exists. However, with the more recent availability of genomic location (ChIP-seq) data, one can approach that question through the identification of characteristic patterns of transcription factor occupancy and histone modifications.
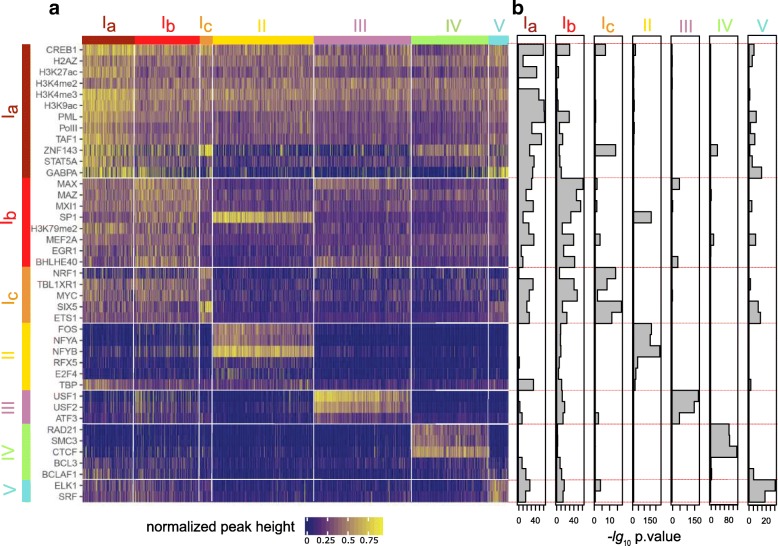
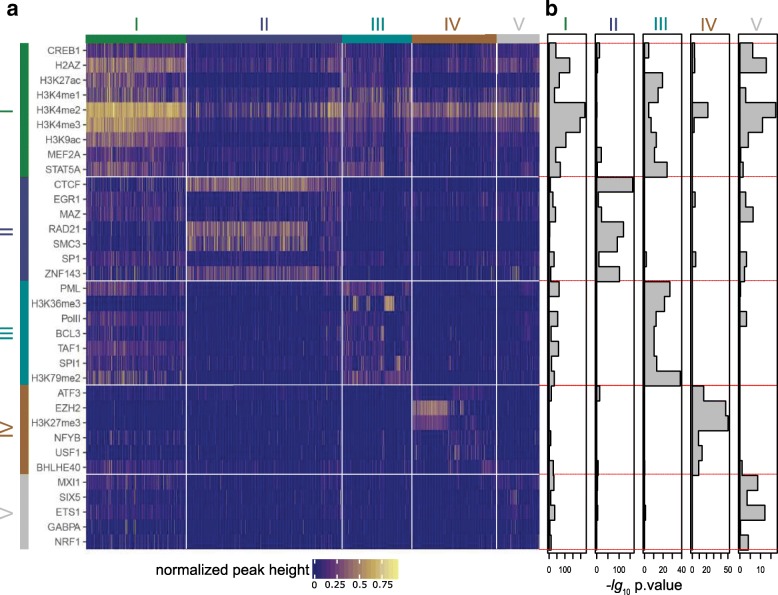
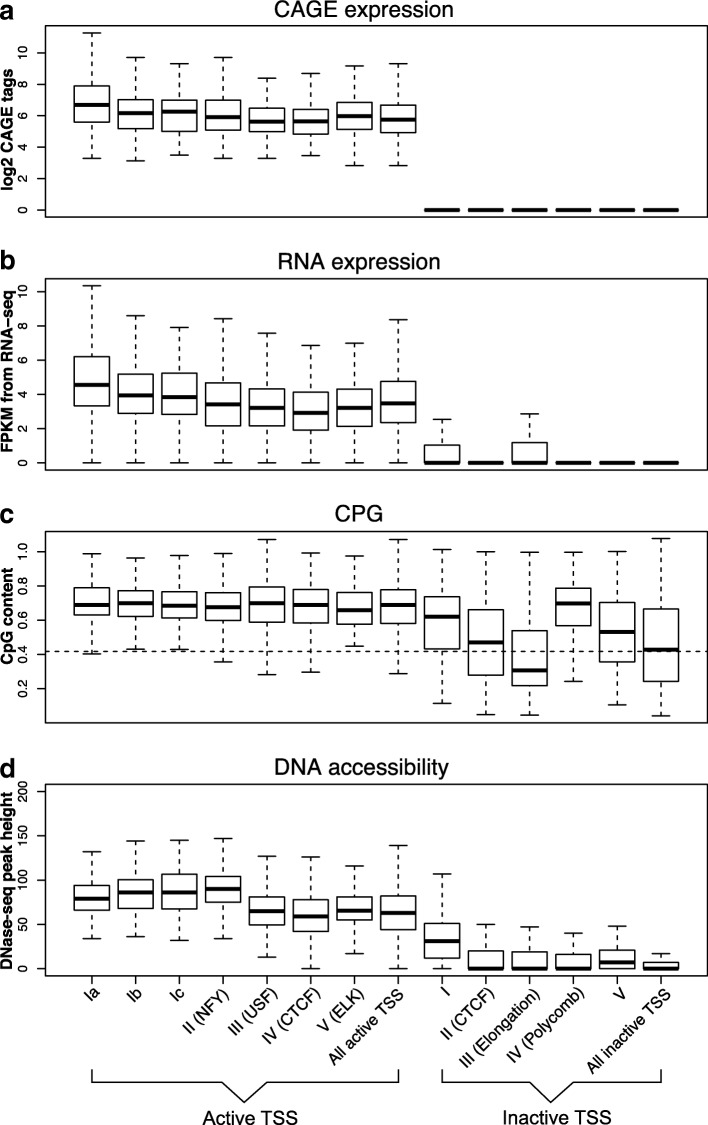
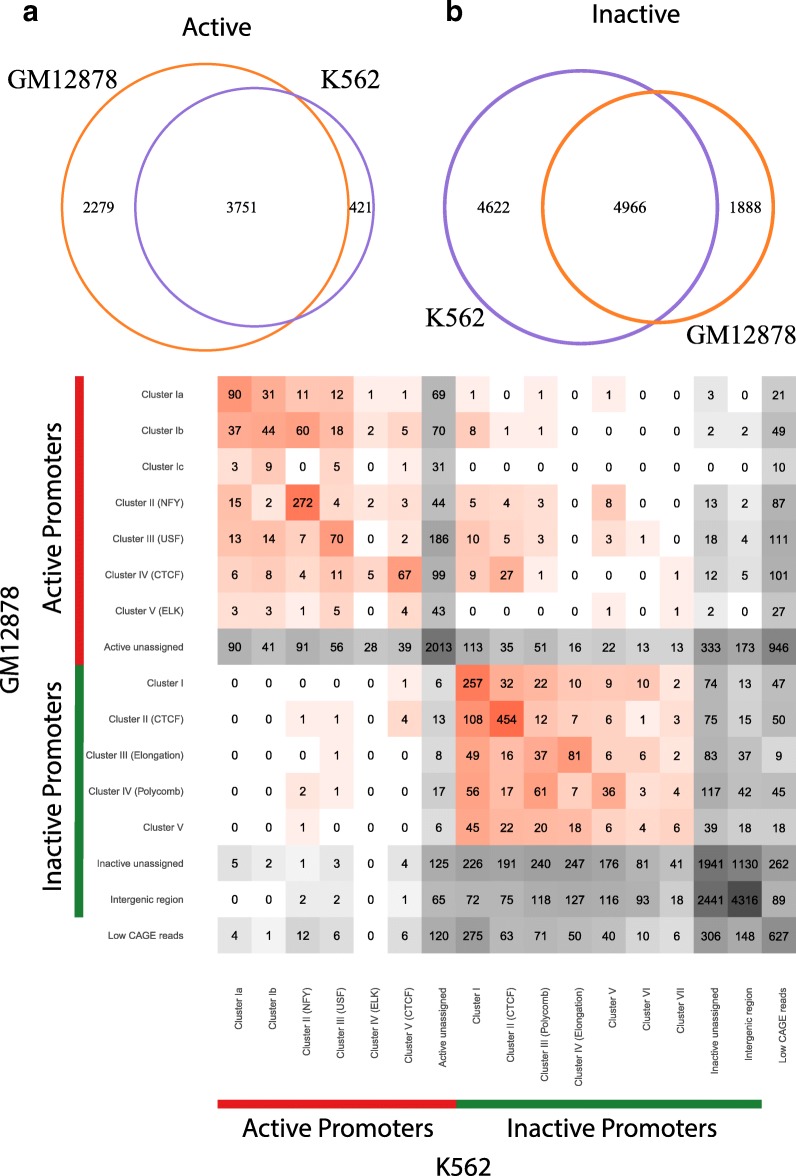
Based on the ENCODE annotation and integration of sequence motifs as well as three-dimensional chromatin data, we have undertaken a re-analysis of occupancy and sequence patterns in human promoters. We identify clear groups of CAAT-box and E-box sequence motif containing promoters, as well as a group of promoters whose interaction with an enhancer appears to be mediated by CCCTC-binding factor (CTCF) binding on the promoter. We also extend our analysis to inactive promoters, showing that only a surprisingly small number of inactive promoters is repressed by the polycomb complex. We also identify combinatorial patterns of transcription factor interactions indicated by the ChIP-seq signals.
Our analysis defines subgroups of promoters characterized by stereotypic patterns of transcription factor occupancy, and combinations of specific sequence patterns which are required for their binding. This grouping provides new hypotheses concerning the assembly and dynamics of transcription factor complexes at their respective promoter groups, as well as questions on the evolutionary origin of these groups.
即使在如今已经几乎完整地了解了启动子的情况下,对人类启动子中的重复序列模式进行特征化描述仍然是一项具有挑战性的任务。然而,随着最近基因组位置(ChIP-seq)数据的可用性,人们可以通过识别转录因子占据和组蛋白修饰的特征模式来解决这个问题。
基于 ENCODE 注释以及序列基序和三维染色质数据的整合,我们对人类启动子中的占据和序列模式进行了重新分析。我们确定了明确的 CAAT 框和 E 盒序列基序含有的启动子组,以及一组似乎通过启动子上的结合因子(CTCF)结合来与增强子相互作用的启动子。我们还将分析扩展到非活性启动子,表明只有一小部分非活性启动子被多梳复合物抑制。我们还通过 ChIP-seq 信号识别出转录因子相互作用的组合模式。
我们的分析定义了启动子的亚组,这些亚组的特征是转录因子占据的刻板模式,以及结合所需的特定序列模式的组合。这种分组为在各自的启动子组中组装和动态转录因子复合物提供了新的假设,以及这些组的进化起源的问题。