Galinsky Kevin J, Reshef Yakir A, Finucane Hilary K, Loh Po-Ru, Zaitlen Noah, Patterson Nick J, Brown Brielin C, Price Alkes L
Department of Biostatistics, Harvard School of Public Health, Boston, Massachusetts.
Takeda Oncology, Cambridge, Massachusetts.
Genet Epidemiol. 2019 Mar;43(2):180-188. doi: 10.1002/gepi.22173. Epub 2018 Nov 25.
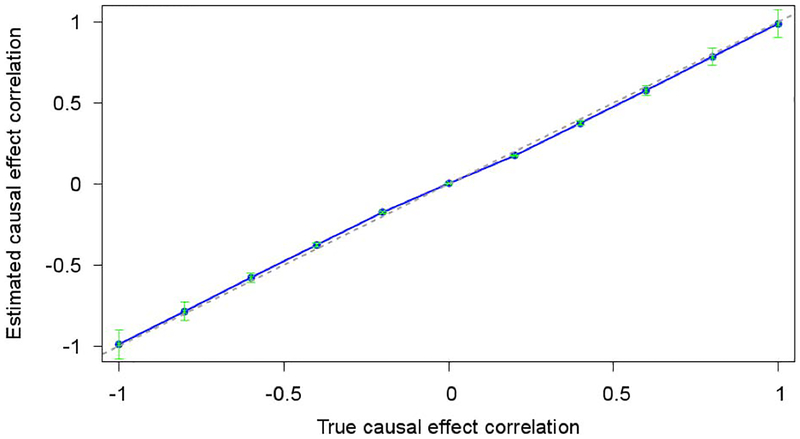
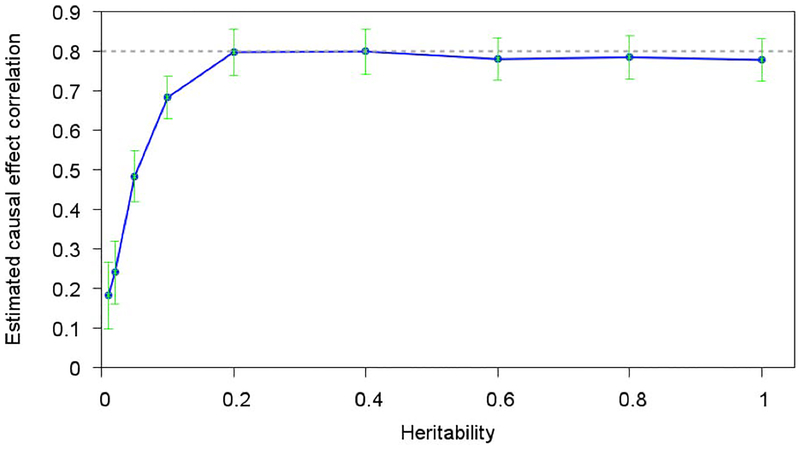
Recent studies have examined the genetic correlations of single-nucleotide polymorphism (SNP) effect sizes across pairs of populations to better understand the genetic architectures of complex traits. These studies have estimated , the cross-population correlation of joint-fit effect sizes at genotyped SNPs. However, the value of depends both on the cross-population correlation of true causal effect sizes ( ) and on the similarity in linkage disequilibrium (LD) patterns in the two populations, which drive tagging effects. Here, we derive the value of the ratio as a function of LD in each population. By applying existing methods to obtain estimates of , we can use this ratio to estimate . Our estimates of were equal to 0.55 ( SE = 0.14) between Europeans and East Asians averaged across nine traits in the Genetic Epidemiology Research on Adult Health and Aging data set, 0.54 ( SE = 0.18) between Europeans and South Asians averaged across 13 traits in the UK Biobank data set, and 0.48 ( SE = 0.06) and 0.65 ( SE = 0.09) between Europeans and East Asians in summary statistic data sets for type 2 diabetes and rheumatoid arthritis, respectively. These results implicate substantially different causal genetic architectures across continental populations.
最近的研究调查了成对人群中单一核苷酸多态性(SNP)效应大小的遗传相关性,以更好地理解复杂性状的遗传结构。这些研究估计了基因型SNP处联合拟合效应大小的跨人群相关性 。然而, 的值既取决于真实因果效应大小的跨人群相关性( ),也取决于驱动标签效应的两个人群中连锁不平衡(LD)模式的相似性。在这里,我们推导出比率 的值作为每个人群中LD的函数。通过应用现有方法获得 的估计值,我们可以使用这个比率来估计 。在成人健康与衰老遗传流行病学数据集中,欧洲人和东亚人之间的 估计值在九个性状上平均为0.55(标准误 = 0.14);在英国生物银行数据集中,欧洲人和南亚人之间的 估计值在13个性状上平均为0.54(标准误 = 0.18);在2型糖尿病和类风湿性关节炎的汇总统计数据集中,欧洲人和东亚人之间的 估计值分别为0.48(标准误 = 0.06)和0.65(标准误 = 0.09)。这些结果表明不同大陆人群之间存在显著不同的因果遗传结构。