Department of Pathology and Laboratory Medicine, David Geffen School of Medicine, University of California, Los Angeles, Los Angeles, CA, USA.
College of Computer Science and Technology, Zhejiang University, Hangzhou, China.
Nat Genet. 2019 Aug;51(8):1244-1251. doi: 10.1038/s41588-019-0465-0. Epub 2019 Jul 29.
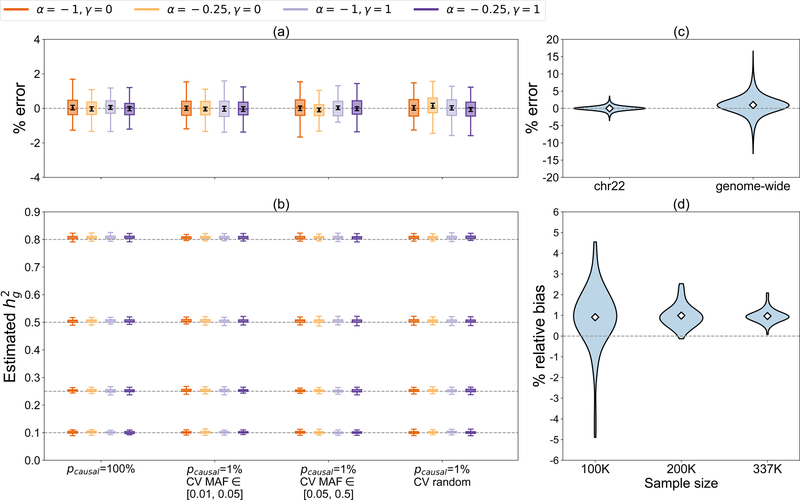
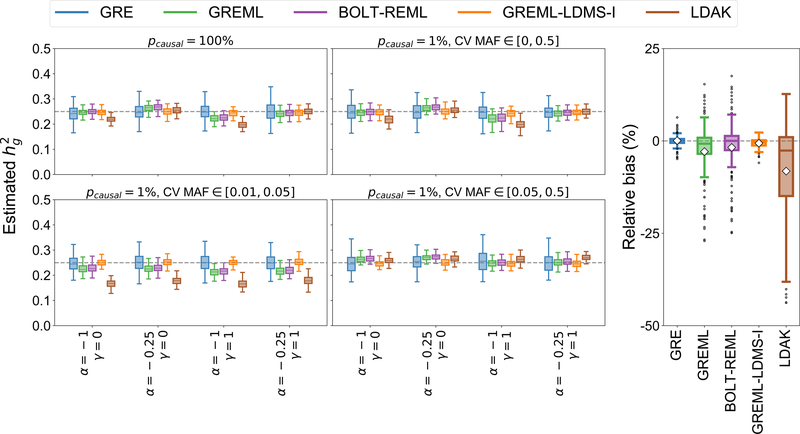
SNP-heritability is a fundamental quantity in the study of complex traits. Recent studies have shown that existing methods to estimate genome-wide SNP-heritability can yield biases when their assumptions are violated. While various approaches have been proposed to account for frequency- and linkage disequilibrium (LD)-dependent genetic architectures, it remains unclear which estimates reported in the literature are reliable. Here we show that genome-wide SNP-heritability can be accurately estimated from biobank-scale data irrespective of genetic architecture, without specifying a heritability model or partitioning SNPs by allele frequency and/or LD. We show analytically and through extensive simulations starting from real genotypes (UK Biobank, N = 337 K) that, unlike existing methods, our closed-form estimator is robust across a wide range of architectures. We provide estimates of SNP-heritability for 22 complex traits in the UK Biobank and show that, consistent with our results in simulations, existing biobank-scale methods yield estimates up to 30% different from our theoretically-justified approach.
SNP 遗传力是研究复杂性状的基本数量。最近的研究表明,当现有方法的假设不成立时,估计全基因组 SNP 遗传力可能会产生偏差。虽然已经提出了各种方法来解释与频率和连锁不平衡(LD)相关的遗传结构,但仍不清楚文献中报告的哪些估计是可靠的。在这里,我们表明,无论遗传结构如何,都可以从生物库规模的数据中准确估计全基因组 SNP 遗传力,而无需指定遗传力模型或根据等位基因频率和/或 LD 对 SNP 进行分区。我们通过从真实基因型(英国生物库,N = 337K)出发的分析和广泛的模拟表明,与现有方法不同,我们的闭式估计器在广泛的结构范围内具有稳健性。我们提供了英国生物库中 22 种复杂性状的 SNP 遗传力估计值,并表明与我们在模拟中的结果一致,现有的生物库规模方法得出的估计值与我们从理论上合理的方法相差高达 30%。