Speck-Planche Alejandro
Research Program on Biomedical Informatics (GRIB), Hospital del Mar Medical Research Institute (IMIM), 08003 Barcelona, Spain.
ACS Omega. 2018 Nov 30;3(11):14704-14716. doi: 10.1021/acsomega.8b02419. Epub 2018 Nov 2.
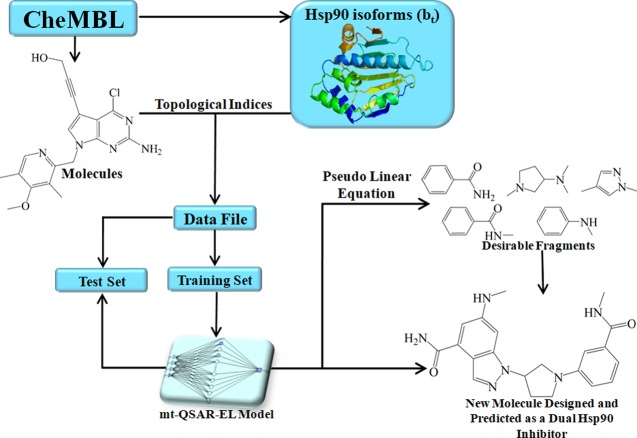
Machine learning methods have revolutionized modern science, providing fast and accurate solutions to multiple problems. However, they are commonly treated as "black boxes". Therefore, in important scientific fields such as medicinal chemistry and drug discovery, machine learning methods are restricted almost exclusively to the task of performing predictions of large and heterogeneous data sets of chemicals. The lack of interpretability prevents the full exploitation of the machine learning models as generators of new chemical knowledge. This work focuses on the development of an ensemble learning model for the prediction and design of potent dual heat shock protein 90 (Hsp90) inhibitors. The model displays accuracy higher than 80% in both training and test sets. To use the ensemble model as a generator of new chemical knowledge, three steps were followed. First, a physicochemical and/or structural interpretation was provided for each molecular descriptor present in the ensemble learning model. Second, the term "pseudolinear equation" was introduced within the context of machine learning to calculate the relative quantitative contributions of different molecular fragments to the inhibitory activity against the two Hsp90 isoforms studied here. Finally, by assembling the fragments with positive contributions, new molecules were designed, being predicted as potent Hsp90 inhibitors. According to Lipinski's rule of five, the designed molecules were found to exhibit potentially good oral bioavailability, a primordial property that chemicals must have to pass early stages in drug discovery. The present approach based on the combination of ensemble learning and fragment-based topological design holds great promise in drug discovery, and it can be adapted and applied to many different scientific disciplines.
机器学习方法彻底改变了现代科学,为多种问题提供了快速准确的解决方案。然而,它们通常被视为“黑匣子”。因此,在药物化学和药物发现等重要科学领域,机器学习方法几乎仅局限于对大量异构化学数据集进行预测的任务。缺乏可解释性阻碍了将机器学习模型充分用作新化学知识的生成器。这项工作专注于开发一种用于预测和设计强效双热休克蛋白90(Hsp90)抑制剂的集成学习模型。该模型在训练集和测试集中的准确率均高于80%。为了将集成模型用作新化学知识的生成器,我们采取了三个步骤。首先,对集成学习模型中存在的每个分子描述符进行了物理化学和/或结构解释。其次,在机器学习的背景下引入了“伪线性方程”,以计算不同分子片段对本文研究的两种Hsp90亚型抑制活性的相对定量贡献。最后,通过组装具有正贡献的片段,设计了新分子,并被预测为强效Hsp90抑制剂。根据Lipinski的五规则,发现所设计的分子具有潜在良好的口服生物利用度,这是化学物质在药物发现早期阶段必须具备的首要特性。基于集成学习和基于片段的拓扑设计相结合的本方法在药物发现中具有很大的前景,并且可以适用于许多不同的科学学科。