Department of Veterinary and Animal Sciences, Faculty of Health and Medical Sciences, University of Copenhagen, DK-1870, Frederiksberg C, Denmark.
Department of Animal Science, Faculty of Agriculture, Universiti Putra Malaysia, UPM, 43400, Serdang, Selangor, Malaysia.
BMC Bioinformatics. 2018 Dec 17;19(1):513. doi: 10.1186/s12859-018-2553-z.
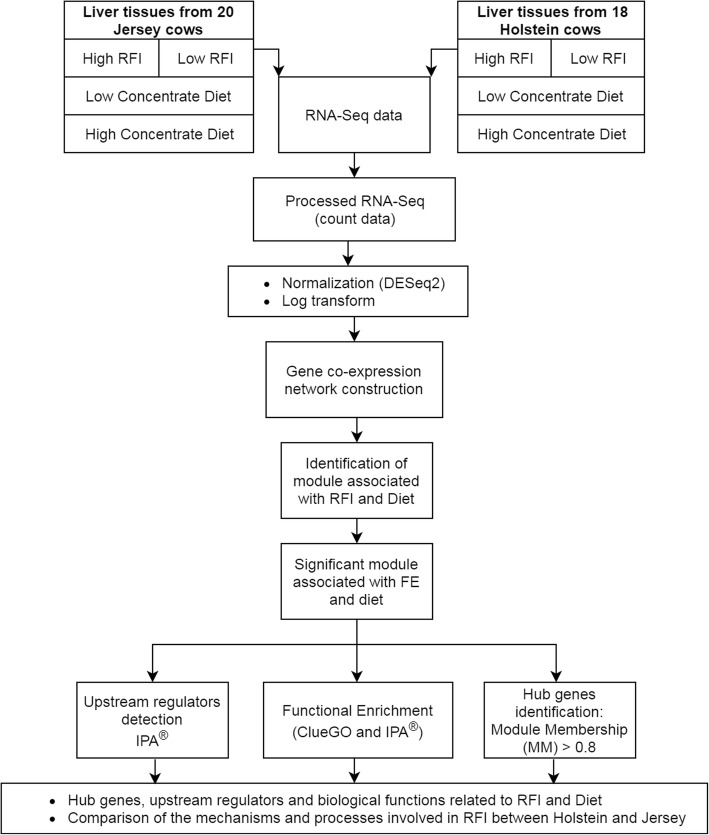
Selection for feed efficiency is crucial for overall profitability and sustainability in dairy cattle production. Key regulator genes and genetic markers derived from co-expression networks underlying feed efficiency could be included in the genomic selection of the best cows. The present study identified co-expression networks associated with high and low feed efficiency and their regulator genes in Danish Holstein and Jersey cows. RNA-sequencing data from Holstein and Jersey cows with high and low residual feed intake (RFI) and treated with two diets (low and high concentrate) were used. Approximately 26 million and 25 million pair reads were mapped to bovine reference genome for Jersey and Holstein breed, respectively. Subsequently, the gene count expressions data were analysed using a Weighted Gene Co-expression Network Analysis (WGCNA) approach. Functional enrichment analysis from Ingenuity® Pathway Analysis (IPA®), ClueGO application and STRING of these modules was performed to identify relevant biological pathways and regulatory genes.
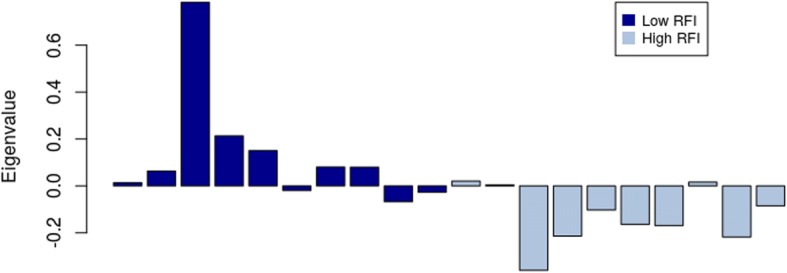
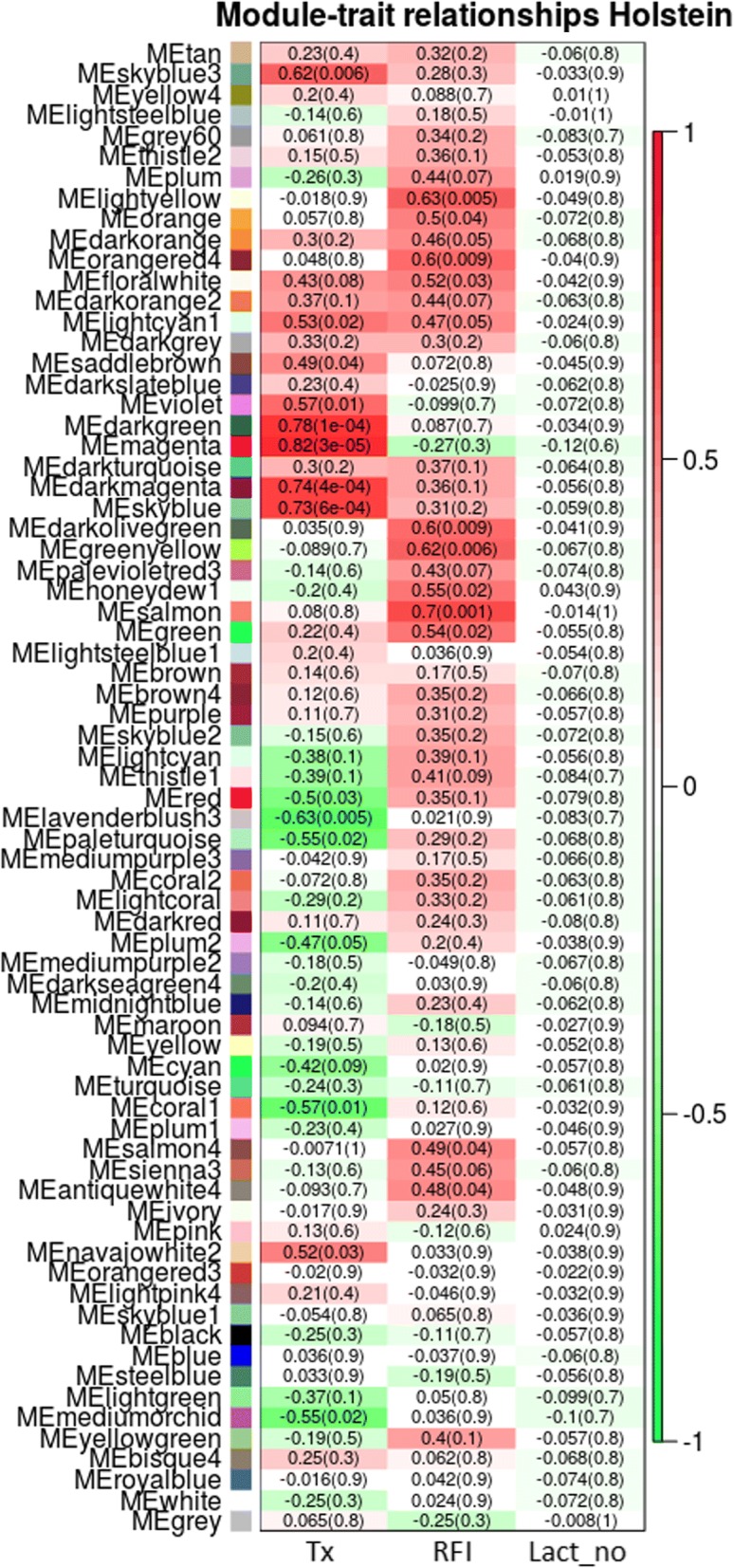
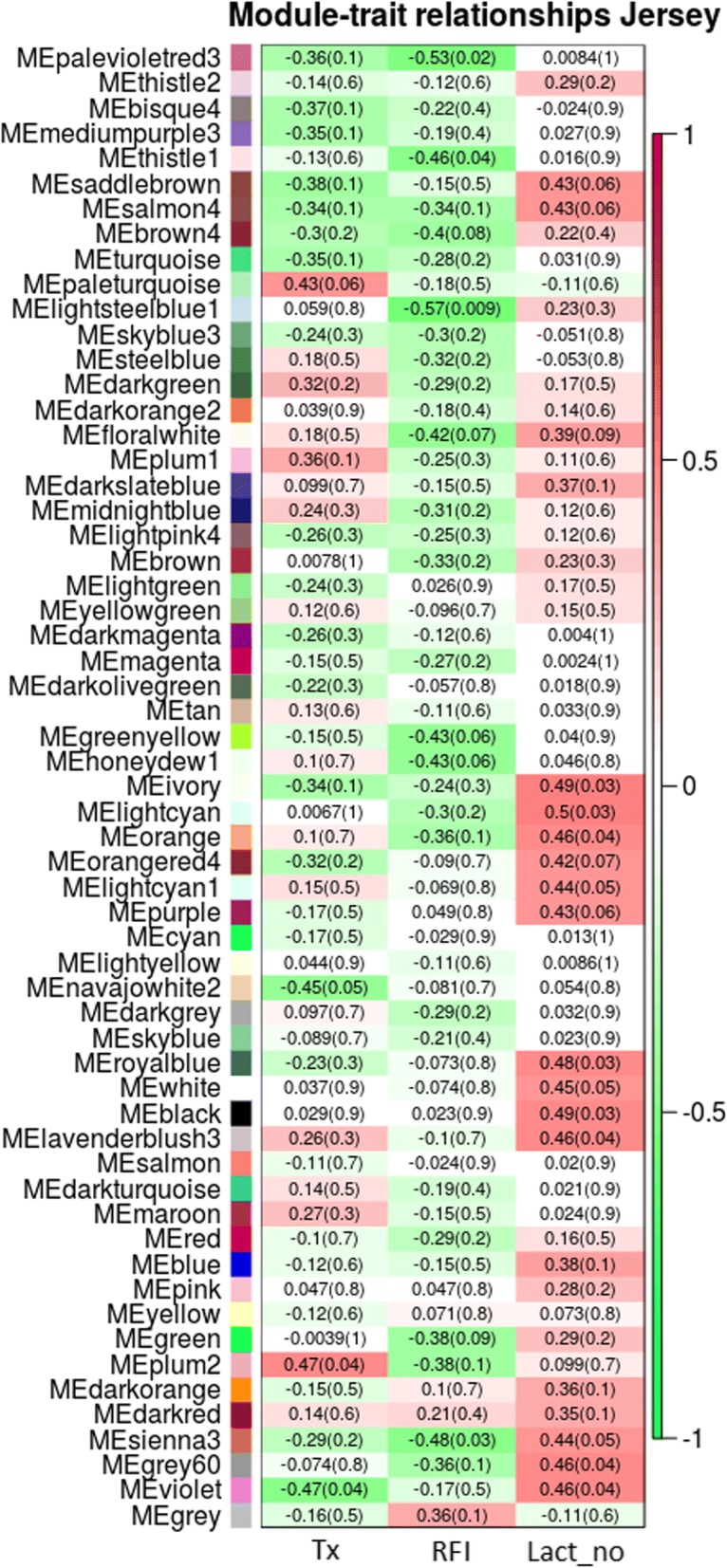
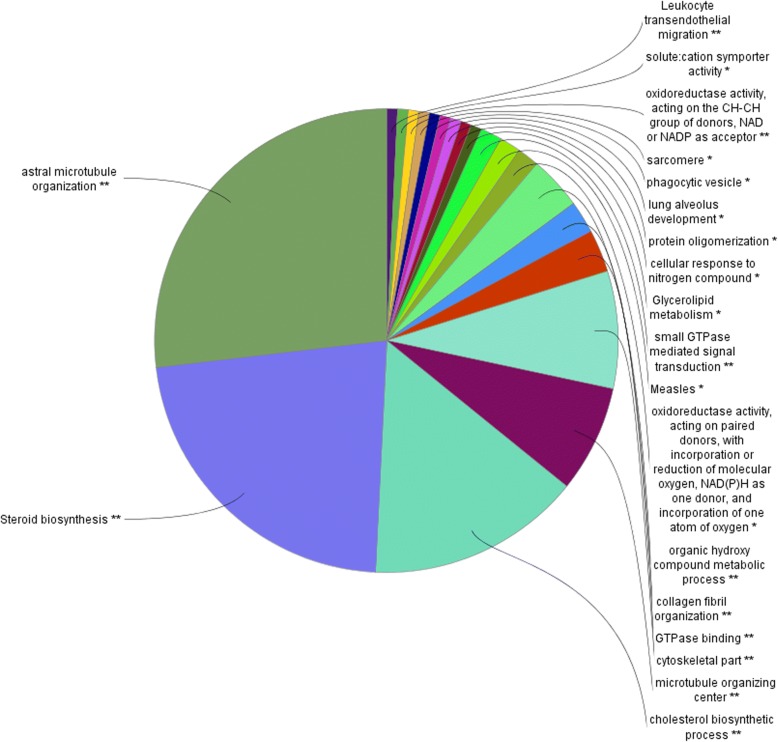
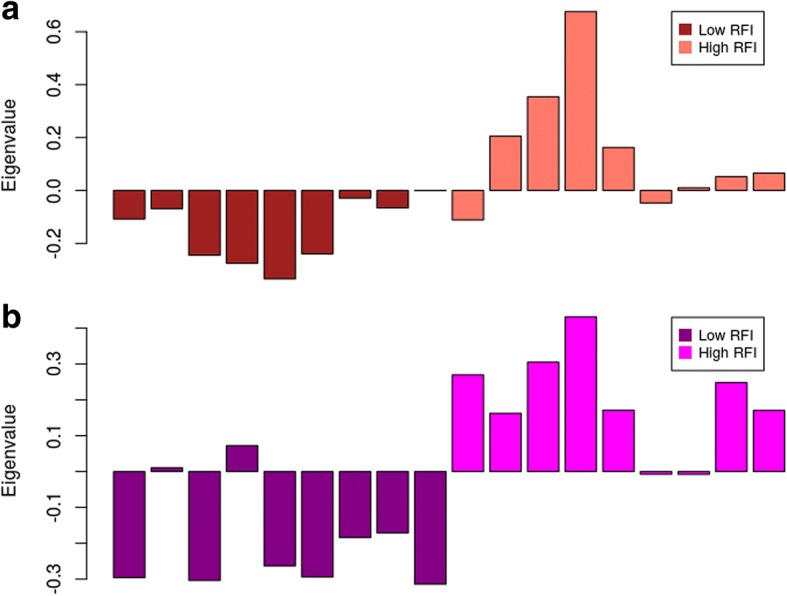
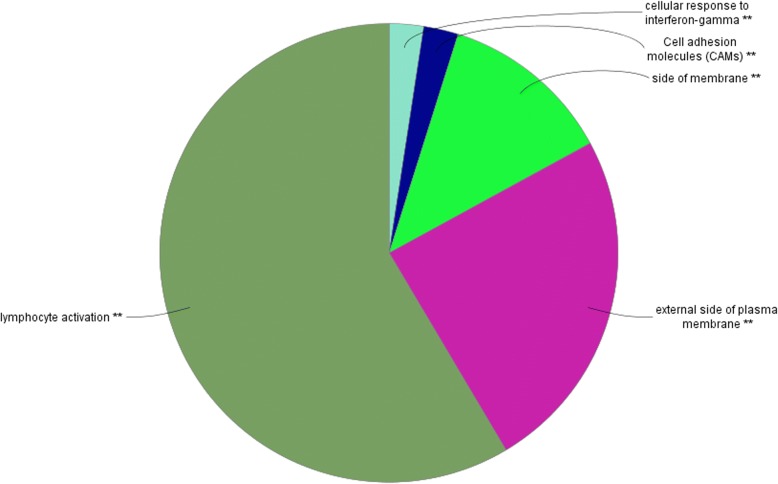
WGCNA identified two groups of co-expressed genes (modules) significantly associated with RFI and one module significantly associated with diet. In Holstein cows, the salmon module with module trait relationship (MTR) = 0.7 and the top upstream regulators ATP7B were involved in cholesterol biosynthesis, steroid biosynthesis, lipid biosynthesis and fatty acid metabolism. The magenta module has been significantly associated (MTR = 0.51) with the treatment diet involved in the triglyceride homeostasis. In Jersey cows, the lightsteelblue1 (MTR = - 0.57) module controlled by IFNG and IL10RA was involved in the positive regulation of interferon-gamma production, lymphocyte differentiation, natural killer cell-mediated cytotoxicity and primary immunodeficiency.
The present study provides new information on the biological functions in liver that are potentially involved in controlling feed efficiency. The hub genes and upstream regulators (ATP7b, IFNG and IL10RA) involved in these functions are potential candidate genes for the development of new biomarkers. However, the hub genes, upstream regulators and pathways involved in the co-expressed networks were different in both breeds. Hence, additional studies are required to investigate and confirm these findings prior to their use as candidate genes.
选择饲料效率对于奶牛生产的整体盈利能力和可持续性至关重要。可以将与饲料效率相关的共表达网络的关键调节基因和遗传标记纳入最佳奶牛的基因组选择中。本研究鉴定了与丹麦荷斯坦牛和泽西牛的高饲料效率和低饲料效率相关的共表达网络及其调节基因。使用了具有高和低残留饲料摄入量(RFI)并接受两种饮食(低和高浓缩物)处理的荷斯坦牛和泽西牛的 RNA-seq 数据。大约 2600 万和 2500 万对读取被映射到荷斯坦牛和泽西牛的牛参考基因组上。随后,使用加权基因共表达网络分析(WGCNA)方法分析基因计数表达数据。使用 IPA®、ClueGO 应用程序和 STRING 对这些模块进行功能富集分析,以确定相关的生物学途径和调节基因。
WGCNA 鉴定了与 RFI 显著相关的两组共表达基因(模块)和一个与饮食显著相关的模块。在荷斯坦牛中,与模块特征关系(MTR)= 0.7 的鲑鱼模块和顶级上游调节因子 ATP7B 参与胆固醇生物合成、类固醇生物合成、脂质生物合成和脂肪酸代谢。与处理饮食显著相关(MTR = 0.51)的紫红色模块参与了甘油三酯稳态。在泽西牛中,lightsteelblue1 模块(MTR = -0.57)由 IFNG 和 IL10RA 控制,涉及干扰素-γ产生、淋巴细胞分化、自然杀伤细胞介导的细胞毒性和原发性免疫缺陷的正调控。
本研究提供了有关肝脏中潜在参与控制饲料效率的生物学功能的新信息。涉及这些功能的枢纽基因和上游调节因子(ATP7b、IFNG 和 IL10RA)是开发新生物标志物的潜在候选基因。然而,这两个品种的共表达网络中涉及的枢纽基因、上游调节因子和途径不同。因此,需要进一步研究以验证这些发现,然后才能将其用作候选基因。