Sorbonne Universités, UPMC, Univ Paris 06 UMRS 1127, INSERM U 1127, CNRS UMR 7225, ICM (Brain and Spine Institute) Pitié-Salpêtrière Hospital, 75013 Paris, France.
Ecole Pratique des Hautes Etudes (EPHE), Paris Sciences et Lettres (PSL) Research University, Neurogenetics Group, 75013 Paris, France.
Dis Model Mech. 2019 Jan 11;12(1):dmm036145. doi: 10.1242/dmm.036145.
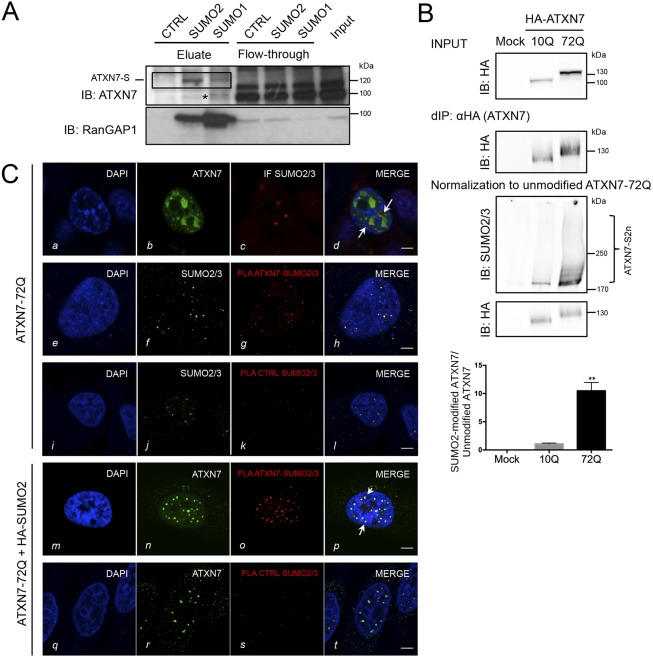
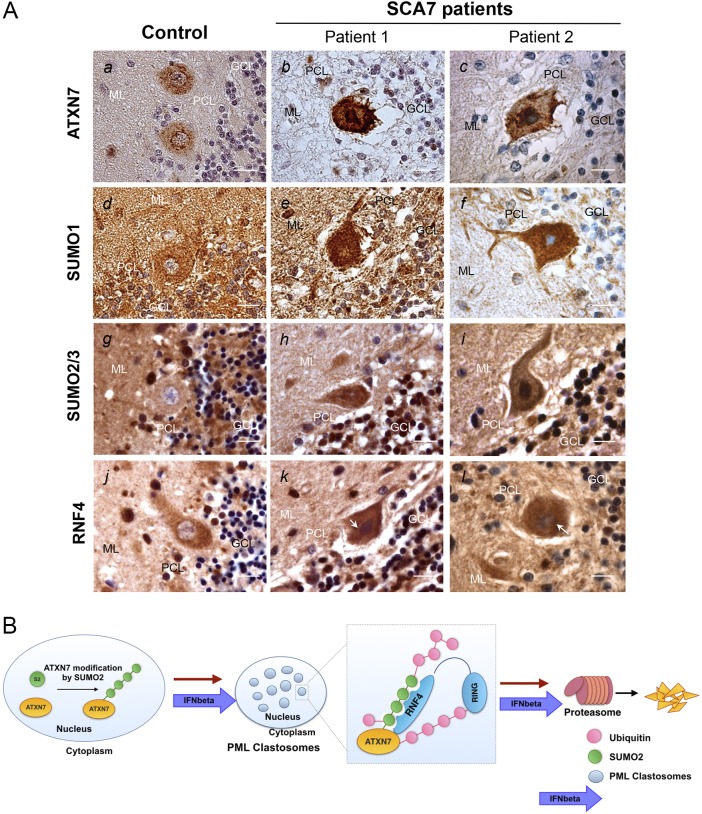
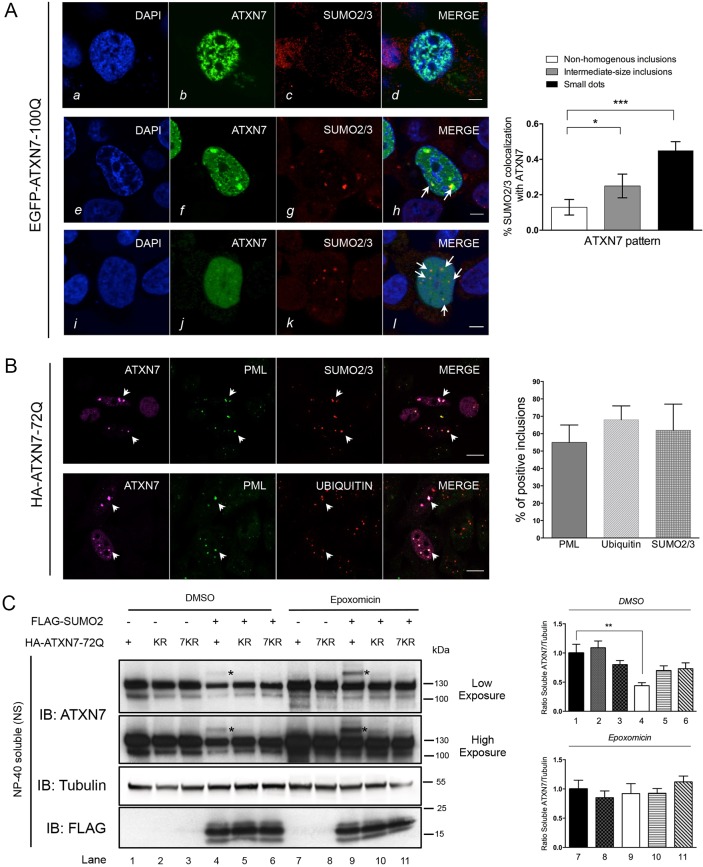
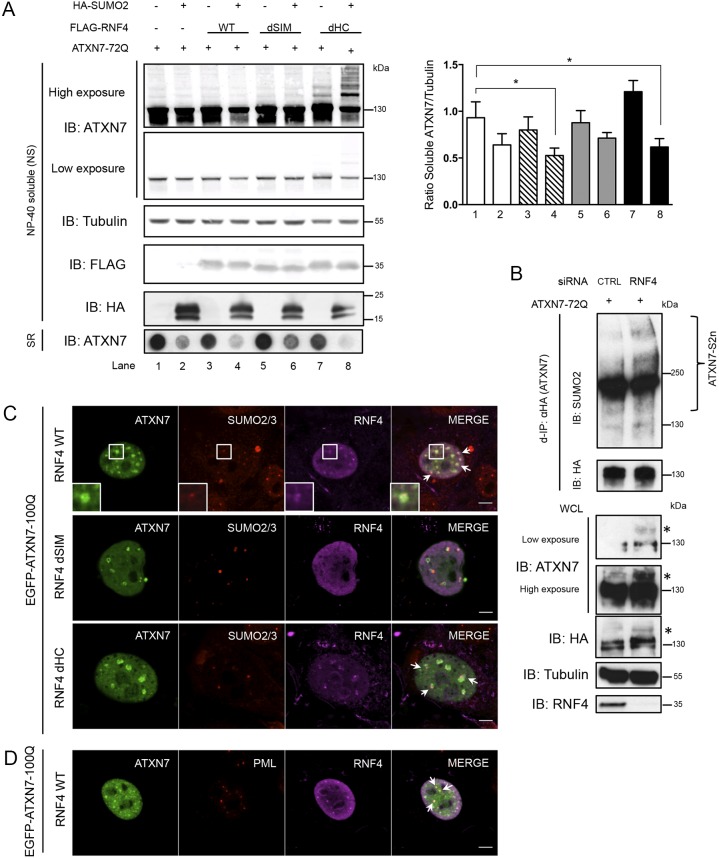
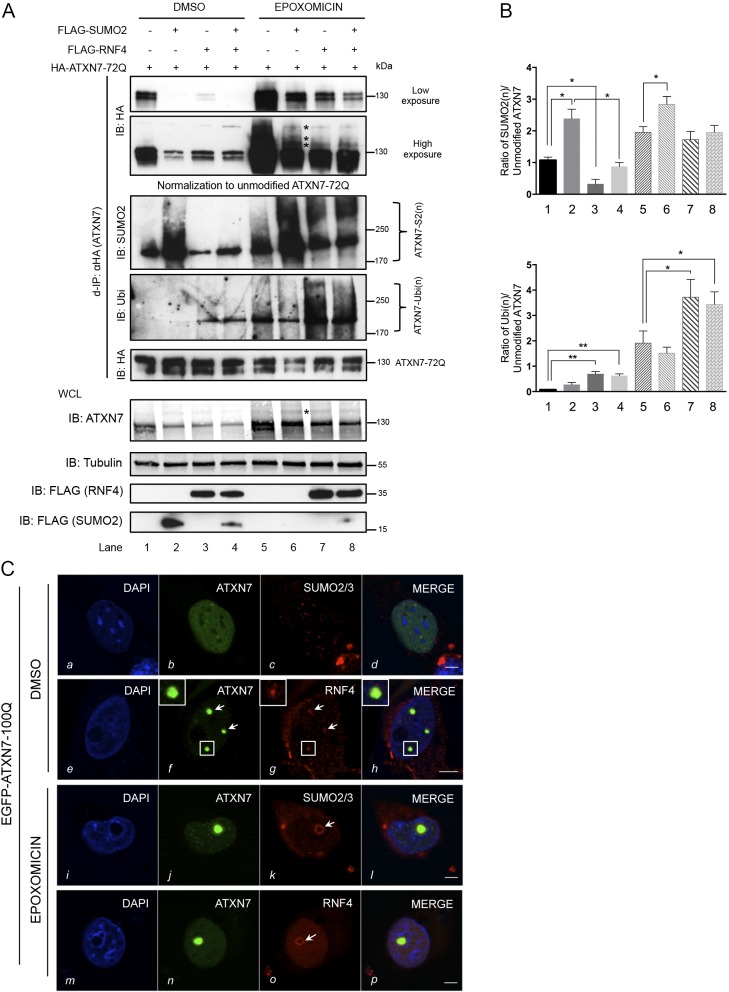
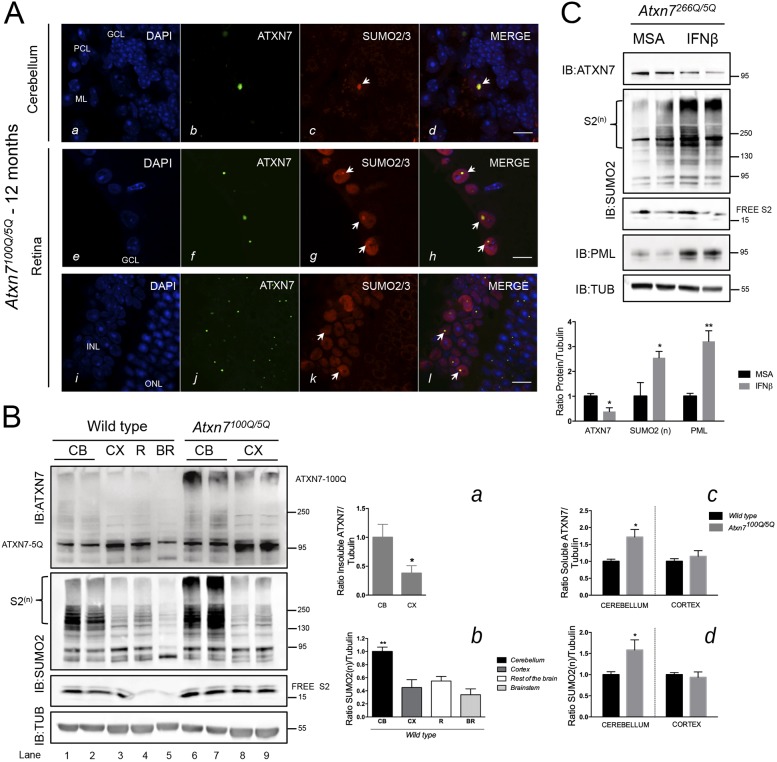
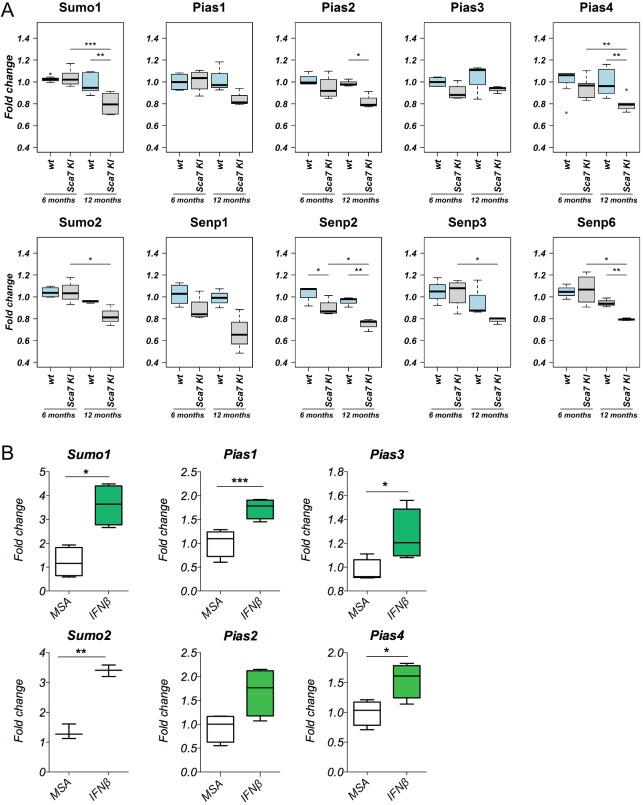
Perturbation of protein homeostasis and aggregation of misfolded proteins is a major cause of many human diseases. A hallmark of the neurodegenerative disease spinocerebellar ataxia type 7 (SCA7) is the intranuclear accumulation of mutant, misfolded ataxin-7 (polyQ-ATXN7). Here, we show that endogenous ATXN7 is modified by SUMO proteins, thus also suggesting a physiological role for this modification under conditions of proteotoxic stress caused by the accumulation of polyQ-ATXN7. Co-immunoprecipitation experiments, immunofluorescence microscopy and proximity ligation assays confirmed the colocalization and interaction of polyQ-ATXN7 with SUMO2 in cells. Moreover, upon inhibition of the proteasome, both endogenous SUMO2/3 and the RNF4 ubiquitin ligase surround large polyQ-ATXN7 intranuclear inclusions. Overexpression of RNF4 and/or SUMO2 significantly decreased levels of polyQ-ATXN7 and, upon proteasomal inhibition, led to a marked increase in the polyubiquitination of polyQ-ATXN7. This provides a mechanism for the clearance of polyQ-ATXN7 from affected cells that involves the recruitment of RNF4 by SUMO2/3-modified polyQ-ATXN7, thus leading to its ubiquitination and proteasomal degradation. In a SCA7 knock-in mouse model, we similarly observed colocalization of SUMO2/3 with polyQ-ATXN7 inclusions in the cerebellum and retina. Furthermore, we detected accumulation of SUMO2/3 high-molecular-mass species in the cerebellum of SCA7 knock-in mice, compared with their wild-type littermates, and changes in SUMO-related transcripts. Immunohistochemical analysis showed the accumulation of SUMO proteins and RNF4 in the cerebellum of SCA7 patients. Taken together, our results show that the SUMO pathway contributes to the clearance of aggregated ATXN7 and suggest that its deregulation might be associated with SCA7 disease progression.
蛋白质平衡的扰乱和错误折叠蛋白的聚集是许多人类疾病的主要原因。神经退行性疾病脊髓小脑共济失调 7 型(SCA7)的一个标志是突变的、错误折叠的 ataxin-7(polyQ-ATXN7)在核内的积累。在这里,我们表明内源性 ATXN7 被 SUMO 蛋白修饰,因此也提示在由 polyQ-ATXN7 积累引起的蛋白毒性应激条件下,这种修饰具有生理作用。共免疫沉淀实验、免疫荧光显微镜和邻近连接分析证实了 polyQ-ATXN7 与 SUMO2 在细胞中的共定位和相互作用。此外,在蛋白酶体抑制后,内源性 SUMO2/3 和 RNF4 泛素连接酶都包围着大的 polyQ-ATXN7 核内包涵体。RNF4 和/或 SUMO2 的过表达显著降低了 polyQ-ATXN7 的水平,并且在蛋白酶体抑制后,导致 polyQ-ATXN7 的多泛素化明显增加。这为涉及 SUMO2/3 修饰的 polyQ-ATXN7 招募 RNF4 的 polyQ-ATXN7 从受影响的细胞中清除提供了一种机制,从而导致其泛素化和蛋白酶体降解。在 SCA7 基因敲入小鼠模型中,我们同样观察到 SUMO2/3 与小脑和视网膜中 polyQ-ATXN7 包涵体的共定位。此外,与野生型同窝仔相比,我们在 SCA7 基因敲入小鼠的小脑检测到 SUMO2/3 高分子量物质的积累,以及 SUMO 相关转录物的变化。免疫组织化学分析显示,SUMO 蛋白和 RNF4 在 SCA7 患者的小脑中有积累。总之,我们的结果表明 SUMO 途径有助于清除聚集的 ATXN7,并提示其失调可能与 SCA7 疾病进展有关。