Brankovics Balázs, Kulik Tomasz, Sawicki Jakub, Bilska Katarzyna, Zhang Hao, de Hoog G Sybren, van der Lee Theo Aj, Waalwijk Cees, van Diepeningen Anne D
Wageningen Plant Research, Wageningen University & Research, Wageningen, Netherlands.
Westerdijk Fungal Biodiversity Institute, Utrecht, Netherlands.
PeerJ. 2018 Dec 19;6:e5963. doi: 10.7717/peerj.5963. eCollection 2018.
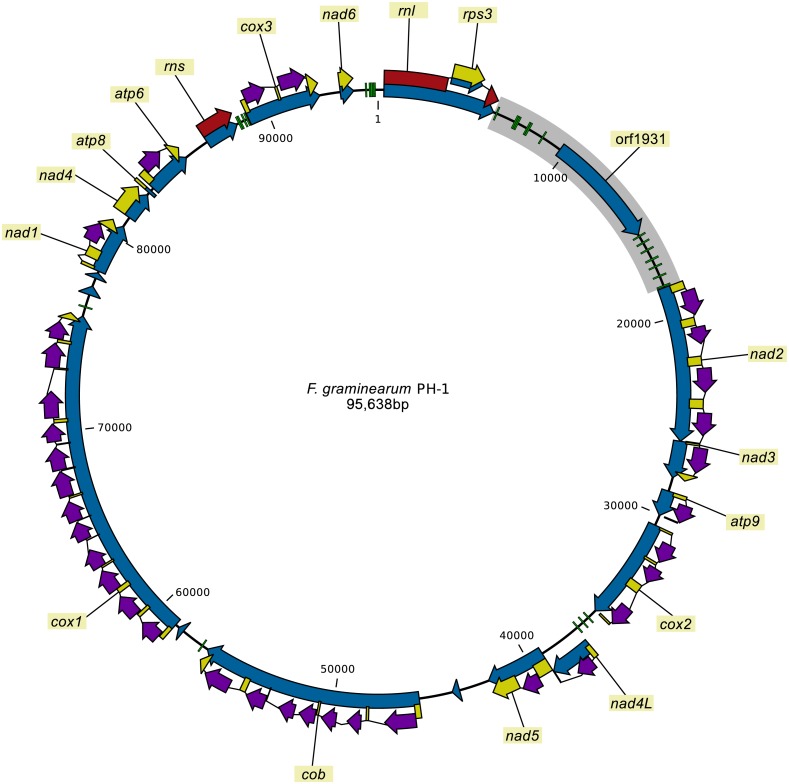
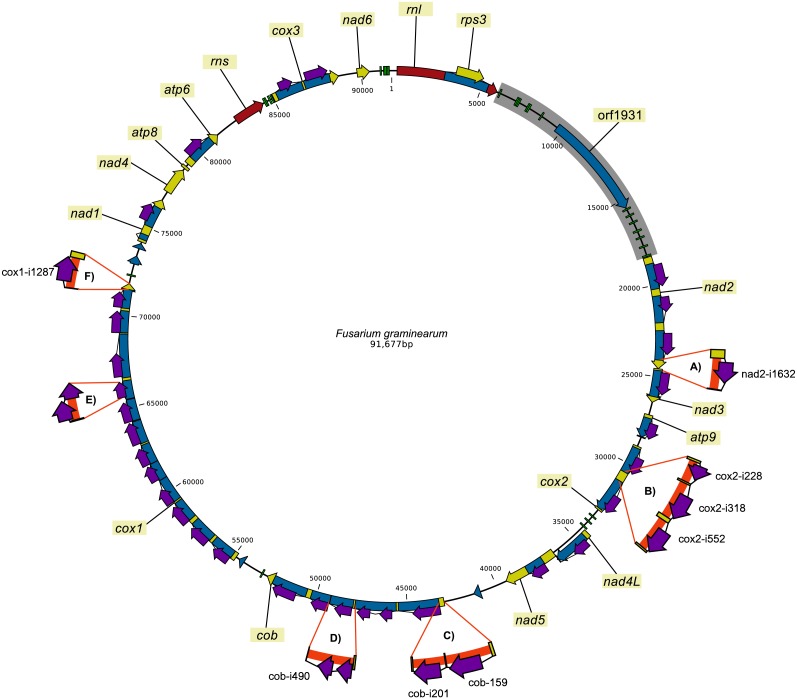
There is a gradual shift from representing a species' genome by a single reference genome sequence to a pan-genome representation. Pan-genomes are the abstract representations of the genomes of all the strains that are present in the population or species. In this study, we employed a pan-genomic approach to analyze the intraspecific mitochondrial genome diversity of . We present an improved reference mitochondrial genome for with an intron-exon annotation that was verified using RNA-seq data. Each of the 24 studied isolates had a distinct mitochondrial sequence. Length variation in the mitogenome was found to be largely due to variation of intron regions (99.98%). The "intronless" mitogenome length was found to be quite stable and could be informative when comparing species. The coding regions showed high conservation, while the variability of intergenic regions was highest. However, the most important variable parts are the intron regions, because they contain approximately half of the variable sites, make up more than half of the mitogenome, and show presence/absence variation. Furthermore, our analyses show that the mitogenome of is recombining, as was previously shown in , indicating that mitogenome recombination is a common phenomenon in . The majority of mitochondrial introns in belongs to group I introns, which are associated with homing endonuclease genes (HEGs). Mitochondrial introns containing HE genes may spread within populations through homing, where the endonuclease recognizes and cleaves the recognition site in the target gene. After cleavage of the "host" gene, it is replaced by the gene copy containing the intron with HEG. We propose to use introns unique to a population for tracking the spread of the given population, because introns can spread through vertical inheritance, recombination as well as via horizontal transfer. We demonstrate how pooled sequencing of strains can be used for mining mitogenome data. The usage of pooled sequencing offers a scalable solution for population analysis and for species level comparisons studies. This study may serve as a basis for future mitochondrial genome variability studies and representations.
从用单个参考基因组序列来代表一个物种的基因组到泛基因组的呈现,这是一个逐渐转变的过程。泛基因组是种群或物种中所有菌株基因组的抽象表示。在本研究中,我们采用泛基因组方法来分析……的种内线粒体基因组多样性。我们为……提供了一个改进的参考线粒体基因组,并带有通过RNA测序数据验证的内含子-外显子注释。所研究的24个分离株中的每一个都有独特的线粒体序列。发现……线粒体基因组的长度变异主要归因于内含子区域的变异(99.98%)。发现“无内含子”的线粒体基因组长度相当稳定,在比较物种时可能具有参考价值。编码区域显示出高度保守性,而基因间区域的变异性最高。然而,最重要的可变部分是内含子区域,因为它们包含大约一半的可变位点,占线粒体基因组的一半以上,并且呈现出存在/缺失变异。此外,我们的分析表明……的线粒体基因组正在发生重组,正如之前在……中所显示的那样,这表明线粒体基因组重组在……中是一种常见现象。……中的大多数线粒体内含子属于I组内含子,它们与归巢内切酶基因(HEGs)相关。含有HE基因的线粒体内含子可能通过归巢在种群中传播,其中内切酶识别并切割靶基因中的识别位点。“宿主”基因被切割后,会被含有带有HEG的内含子的基因拷贝所取代。我们建议使用种群特有的内含子来追踪给定种群的传播,因为内含子可以通过垂直遗传、重组以及水平转移进行传播。我们展示了如何将菌株的混合测序用于挖掘线粒体基因组数据。混合测序的使用为种群分析和物种水平比较研究提供了一种可扩展的解决方案。本研究可为未来线粒体基因组变异性研究和呈现提供基础。