Wang Li, Pang Kaifang, Han Kihoon, Adamski Carolyn J, Wang Wei, He Lingjie, Lai Jason K, Bondar Vitaliy V, Duman Joseph G, Richman Ronald, Tolias Kimberley F, Barth Patrick, Palzkill Timothy, Liu Zhandong, Holder J Lloyd, Zoghbi Huda Y
Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX, 77030, USA.
Jan and Dan Duncan Neurological Research Institute at Texas Children's Hospital, Houston, TX, 77030, USA.
Mol Psychiatry. 2020 Oct;25(10):2534-2555. doi: 10.1038/s41380-018-0324-x. Epub 2019 Jan 4.
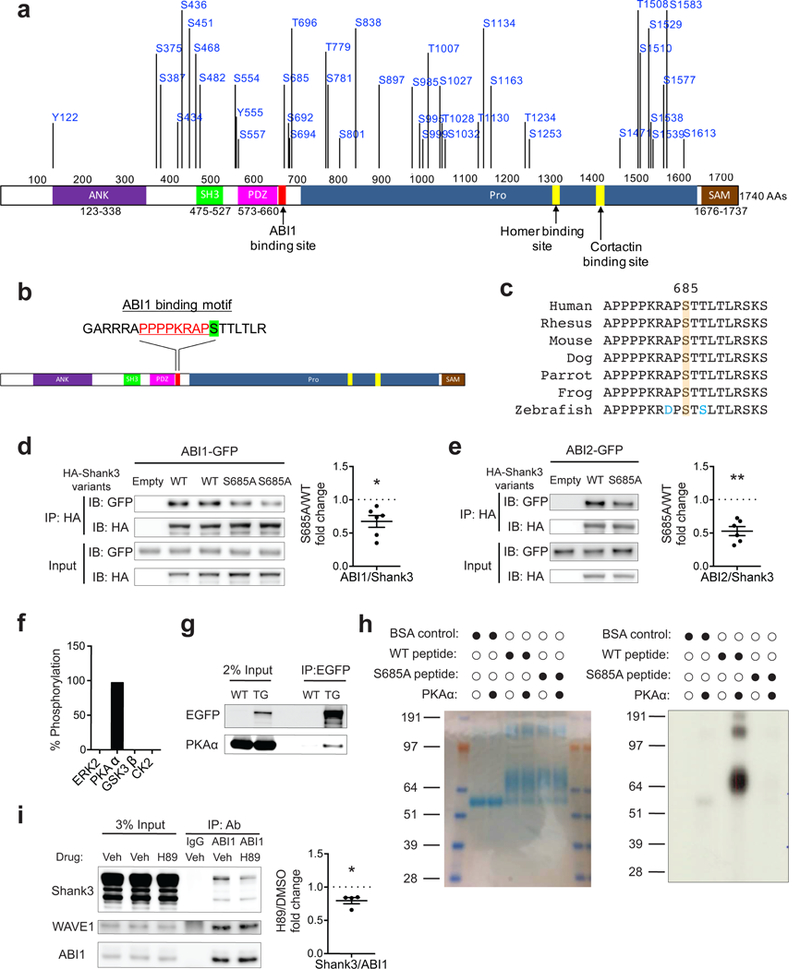
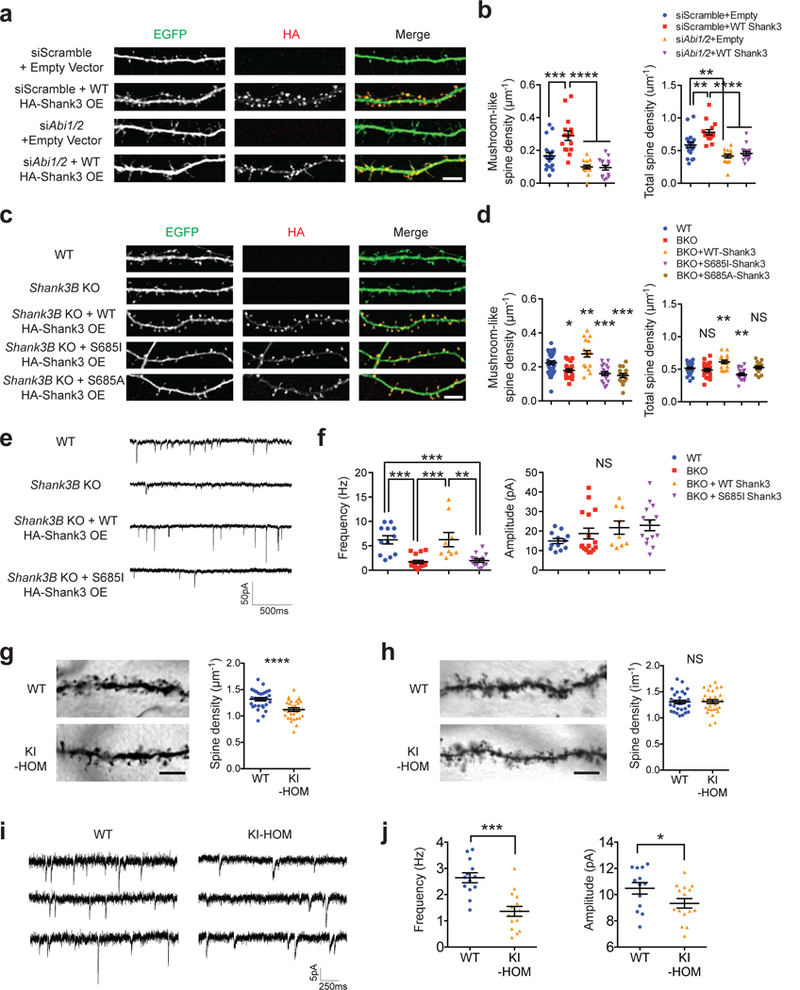
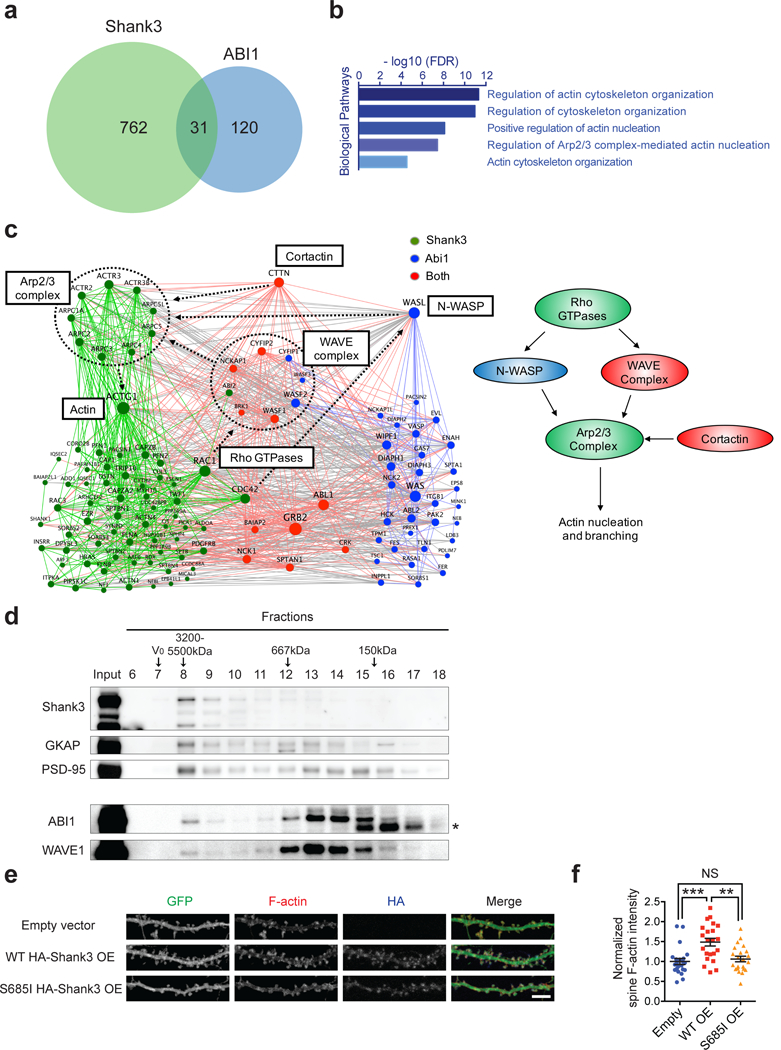
Genome sequencing has revealed an increasing number of genetic variations that are associated with neuropsychiatric disorders. Frequently, studies limit their focus to likely gene-disrupting mutations because they are relatively easy to interpret. Missense variants, instead, have often been undervalued. However, some missense variants can be informative for developing a more profound understanding of disease pathogenesis and ultimately targeted therapies. Here we present an example of this by studying a missense variant in a well-known autism spectrum disorder (ASD) causing gene SHANK3. We analyzed Shank3's in vivo phosphorylation profile and identified S685 as one phosphorylation site where one ASD-linked variant has been reported. Detailed analysis of this variant revealed a novel function of Shank3 in recruiting Abelson interactor 1 (ABI1) and the WAVE complex to the post-synaptic density (PSD), which is critical for synapse and dendritic spine development. This function was found to be independent of Shank3's other functions such as binding to GKAP and Homer. Introduction of this human ASD mutation into mice resulted in a small subset of phenotypes seen previously in constitutive Shank3 knockout mice, including increased allogrooming, increased social dominance, and reduced pup USV. Together, these findings demonstrate the modularity of Shank3 function in vivo. This modularity further indicates that there is more than one independent pathogenic pathway downstream of Shank3 and correcting a single downstream pathway is unlikely to be sufficient for clear clinical improvement. In addition, this study illustrates the value of deep biological analysis of select missense mutations in elucidating the pathogenesis of neuropsychiatric phenotypes.
基因组测序揭示了越来越多与神经精神疾病相关的基因变异。通常,研究将重点局限于可能破坏基因的突变,因为它们相对易于解读。相反,错义变体常常被低估。然而,一些错义变体对于更深入理解疾病发病机制以及最终的靶向治疗可能具有重要意义。在此,我们通过研究一种已知的自闭症谱系障碍(ASD)致病基因SHANK3中的错义变体来举例说明这一点。我们分析了Shank3的体内磷酸化谱,并确定S685为一个磷酸化位点,已有报道一种与ASD相关的变体位于该位点。对该变体的详细分析揭示了Shank3在将阿贝尔逊相互作用蛋白1(ABI1)和WAVE复合物募集到突触后致密区(PSD)方面的新功能,这对突触和树突棘的发育至关重要。发现该功能独立于Shank3的其他功能,如与GKAP和Homer的结合。将这种人类ASD突变引入小鼠后,导致出现了一小部分先前在组成型Shank3基因敲除小鼠中观察到的表型,包括异体梳理行为增加、社会优势增加和幼崽超声波发声减少。总之,这些发现证明了Shank3在体内功能的模块化。这种模块化进一步表明,Shank3下游存在不止一条独立的致病途径,纠正单一的下游途径不太可能足以实现明显的临床改善。此外,本研究说明了对特定错义突变进行深入生物学分析在阐明神经精神表型发病机制方面的价值。